|
目前,结直肠癌(colorectal cancer,CRC)的发病率及死亡率呈逐年增长的趋势[1],而CRC的发生是机体易感性和环境因素共同作用的结果[2]。随着宏基因组学的深入研究和分子生物学技术的发展,肠道定植菌群作为重要环境因素在CRC发生发展过程中的作用研究越来越受到重视。肠道定植菌群是肠道的正常菌群,如类杆菌、乳杆菌、大肠埃希菌和肠球菌等,调节宿主与环境之间处于动态平衡,作为重要的环境因素。肠道定植菌群失调及其代谢产物是诱发CRC的关键因素[3,4]。然而,受到研究方法和条件的限制,目前关于肠道定植菌群在CRC发生发展、诊断及治疗中的作用研究报道较少。因此,本研究采用第二代IlluminaMiseq测序技术检测和分析正常肠黏膜组织和结直肠癌组织标本中的定植菌群结构和组分变化,初步探讨肠黏膜定植菌群是否可以作为CRC早期辅助诊断的标志物。
1 对象与方法 一、对象 研究对象为2016年1月至2017年10月同济大学附属同济医院病理确诊的CRC患者28例,其中男20例,女8例,年龄(56.7±8.4)岁;健康对照组12名,其中男8名,女4名,年龄(49.5±7.6)岁。均在结肠镜下取肠道黏膜活检样本(约1 mm×2 mm)。所有患者在采样前1个月均未使用过抗生素药物。所有志愿者都居住在上海地区,无特殊饮食习惯,采样前无肝、胆、胰腺等消化系统疾病史。以上40份组织用生理盐水冲洗表面,收集于冻存管内,然后放置在能立即速冻的没有防腐剂的液氮后储存在-80 ℃冰箱内。
结直肠癌的诊断依据中国结直肠癌诊疗规范(2015版)[5],结直肠癌TNM分期参照美国癌症联合委员会(AJCC)/国际抗癌联盟(UICC)结直肠癌TNM分期系统(2010年第7版)[5]。本研究CRC患者包括Ⅰ~Ⅱ期21例[男15例,女6例,年龄(55.4±7.9)岁],Ⅲ~Ⅳ期7例[男5例,女2例,年龄(57.2±9.9)岁]。本研究已报同济大学附属同济医院伦理委员会并得到批准[(同)伦审-KYSB-2014-(02)],所有研究对象都填写问卷调查表并签署书面形式的知情同意书。
二、方法 1.DNA抽提和PCR扩增: 根据E.Z.N.A.® Soil试剂盒(Omega Bio-tek,Norcross,GA,USA)说明书进行总DNA抽提,DNA浓度和纯度利用NanoDrop2000进行检测,利用1%琼脂糖凝胶电泳检测DNA提取质量;用338F(5′-ACTCCTACGGGA GGCAGCAG-3′)和806R(5′-GGACTACHVGGGTWTCTAAT-3′)引物对V3-V4可变区进行PCR扩增,扩增程序为:95 ℃预变性3 min,27个循环(95 ℃变性30 s,55 ℃退火30 s,72 ℃延伸30 s),最后72 ℃延伸10 min(PCR仪:ABI GeneAmp® 9700型)。扩增体系为20 μl,4 μl 5×FastPfu缓冲液,2 μl 2.5mmol/L dNTPs,0.8 μl引物(5 μmol/L),0.4 μl FastPfu聚合酶;10 ng DNA模板。
2.Illumina Miseq测序: 使用2%琼脂糖凝胶回收PCR产物,利用AxyPrepDNA Gel Extraction Kit(Axygen Biosciences,Union City,CA,USA)进行纯化,Tris-HCl洗脱,2%琼脂糖电泳检测。利用QuantiFluor™-ST(Promega,USA)进行检测定量。根据Illumina MiSeq平台(Illumina,San Diego,USA)标准操作规程将纯化后的扩增片段构建PE 2×300的文库。构建文库步骤:(1)连接"Y"字形接头;(2)使用磁珠筛选去除接头自连片段;(3)利用PCR扩增进行文库模板的富集;(4)氢氧化钠变性,产生单链DNA片段。利用美国Illumina公司的Miseq PE300平台进行测序,原始数据上传至美国国立生物技术信息中心(Nationalcenter for biotechnology information,NCBI)数据库中(序列号:SRP20170523050)。
3.数据处理: 原始测序序列使用Trimmomatic软件质控,使用FLASH软件进行拼接:(1)设置50 bp的窗口,如果窗口内的平均质量值低于20,从窗口开始截去后端碱基,去除质控后长度低于50 bp的序列;(2)Barcode需精确匹配,引物允许2个碱基的错配,去除模糊碱基;(3)根据重叠碱基overlap将两端序列进行拼接,overlap需大于10 bp,去除无法拼接的序列。
使用的UPARSE软件(version 7.1,http://drive5.com/uparse/),根据97%的相似度对序列进行操作单元(operational taxonomic unit,OTU)聚类;使用UCHIME软件剔除嵌合体;利用RDP Classifier(http://rdp.cme.msu.edu/)对每条序列进行物种分类注释,比对Silva数据库(SSU123),设置比对阈值为70%。
三、统计学方法 采用SPSS 17.0软件进行统计学分析。两样本数据间比较采用Mann-Whitney检验,以P<0.05作为差异有统计学意义。
2 结 果 一、正常肠黏膜组织与结直肠癌组织定植菌群结构在门水平上的比较 在人类肠道定植菌群中,细菌门的群类是相当保守的,可以直接反映出不同组织部位定植菌群结构的异质性。在分析肠道定植菌群组分的时候,门也是最高的分类单元,门的群类是非常保守的,因此应该首先说明细菌门的相对丰度的变化规律。
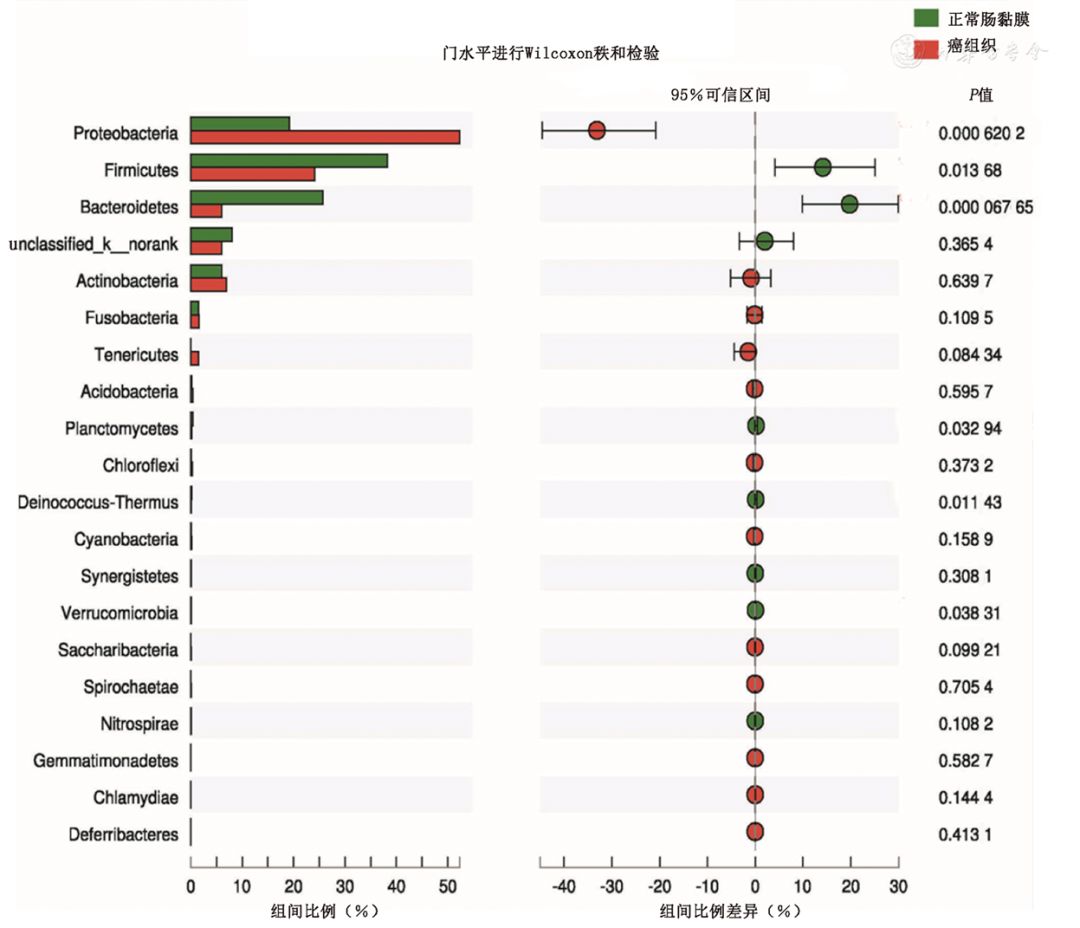
对40份样本鉴定的定植菌群进行组间细菌门水平的比较分析,从图1可以看出,丰度前20位定植菌群大部分在正常肠黏膜和癌组织中附着基本一致,但变形菌门(Proteobacteria)在肠癌组织中显著增加(Z=-1.997,P<0.01),而厚壁菌门(Firmicutes)、拟杆菌门(Bacteroidetes)则在肠癌组织中的丰度显著降低(Z=-1.964,P<0.01),这说明在CRC发生发展过程中定植菌群是动态变化的,这为进一步研究定植菌群在肠癌发生发展中的作用提供基础。
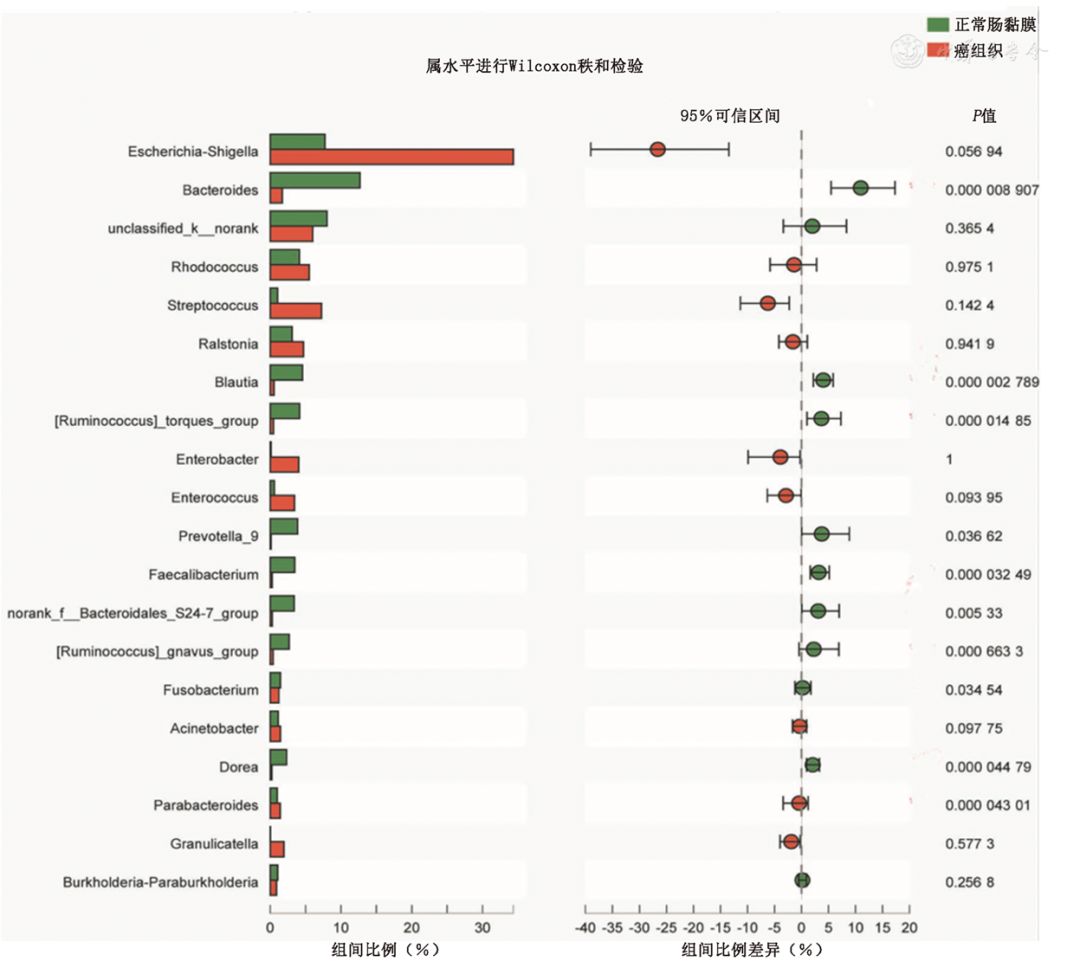
图1 正常肠黏膜与癌组织定植菌群在门水平差异性检验 二、正常肠黏膜组织与结直肠癌组织定植菌群结构在属水平上的比较 在细菌属水平,测序结果显示癌组织有许多菌属明显低于正常肠黏膜组织,其中存在显著性差异的主要菌属为拟杆菌属(Bacteroides)、布劳特菌属(Blautia)、扭链胃球菌属(Ruminococcus torques)、普雷沃菌属(Prevotella)和普拉梭菌(Faecalibacterium)(Z=-3.008,P<0.05);而紫单胞菌科Parabacteroides在癌组织中的丰度比正常肠黏膜组织显著增加,差异有统计学意义(Z=-1.997,P<0.001),如图2所示。
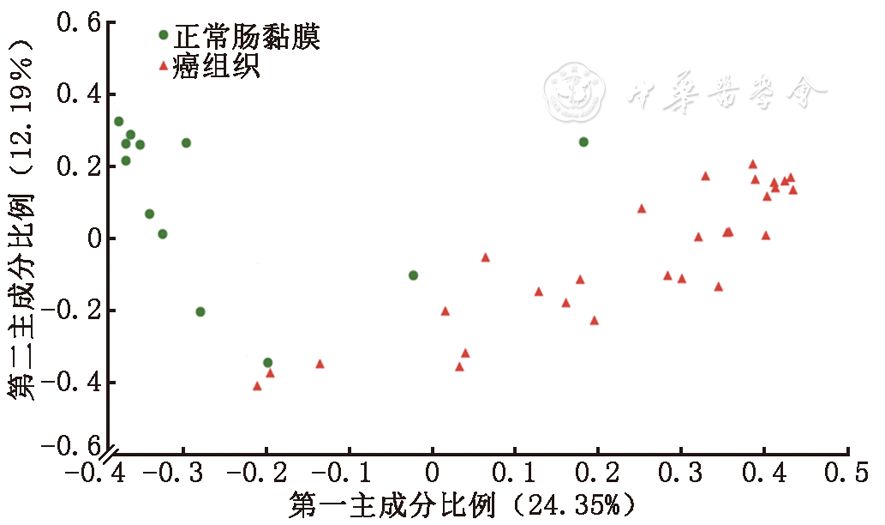
图2 正常肠黏膜与肠癌组织定植菌群在属水平差异性检验 三、正常肠黏膜组织与结直肠癌患者癌组织定植菌群聚类分析 对正常肠黏膜与癌组织之间定植菌群进行了以UniFrac为基础的PCoA聚类分析,从图3中可以看出个体间以疾病的有无进行了明显的聚类,说明在CRC发生以后肠黏膜定植菌群组分及结构发生明显变化,这种明显的聚类与年龄、性别等因素并没有关系。在此基础上,对正常黏膜及肠癌组织定植菌群进行了以偏最小二乘法判别分析(partial least squares discrimination analysis,PLS-DA)为基础的聚类分析,如图4。PLS-DA的聚类进一步证实了在CRC发生以后定植菌群结构、组分与正常肠黏膜具有显著差异。
图3 UniFrac为基础的所有样本的PCoA聚类分析
图4 PLS-DA为基础的所有样本的聚类分析 四、正常肠黏膜组织与结直肠癌组织定植菌群组分分析 在细菌属水平上,测序结果显示有的菌属仅定植于正常肠黏膜组织,其中丰度排前3位的是布赫纳什菌属(Buchnera)、普氏菌属(Prevotella)和普雷沃菌科(Prevotellaceae),分别占48.21%、30.62%和8.91%(图5A);测序结果显示有的菌属仅定植于癌组织,丰度排前3位的是卡氏菌属(Catonella)、库特菌属(Kurthia)和单胞菌属(Brachymonas),分别占7.89%、7.37%和7.33%(图5B)。
图5 正常肠黏膜组织与癌组织定植菌群组分。A图示正常肠粘膜组织仅有的定植菌群(属)及其所占比例;B图示肠癌组织仅有的定植菌群(属)及其所占比例 3 讨 论 结直肠癌的发生是一个多步骤、多途径的过程,更是一个机体内因与环境等外部因素长期交互作用的结果,涉及进行性的分子遗传学改变和相关的组织形态学改变[6]。虽然传统的对于结直肠癌的研究集中在遗传突变和表观遗传的突变上,但随着宏基因组学的发展及二代测序技术的广泛应用,作为重要环境因素的肠道菌群日益受到人们的重视。而且大量的研究表明[7,8,9,10],一些特殊的肠道菌群与CRC的发病机制密切相关,包括牛链球菌、梭状芽胞杆菌、幽门螺旋杆菌等。有些细菌菌株能抑制大肠癌的发生,如嗜酸乳酸杆菌和双歧杆菌[11,12],而有些细菌菌株能促进大肠癌的发生,如毒性脆弱拟杆菌和梭菌属等[13,14]。
目前二代测序技术Illumina Miseq可实现低成本、高通量、高效率的数据产出,大量实例表明基于二代测序技术可揭示特定生存环境中至少80%~90%的微生物种类。因此,在非培养条件下,采用细菌16S rRNA基因(用于细菌分类的保守分子标记)的PCR扩增和扩増产物的高通量测序,理论上能够全面系统揭示CRC不同发展阶段的肠道菌群结构和组分变化。肠道黏膜组织作为样本具有部位准确,能够客观显示病变部位以及健康肠道部位肠道菌群结构与组分,使肠道菌群和CRC发生发展的关系得到进一步揭示,许多研究结果也提供了强力支持。Marchesi[15]通过比较6个结直肠癌患者癌变组织和癌旁组织黏附细菌的群落结构,发现癌变组织富含Coriobacteria类细菌(放线菌门,在正常人的肠道内属于稀有类群),提示了放线菌门细菌成员在癌组织上过度生长与结直肠癌的发生可能相关。另有研究报道[16,17],定植在癌组织上的肠道核梭杆菌(Fusobacteriumnucleatum)(在正常人的肠道内属于稀有类群)引发的感染可能与CRC的发生密切相关。Arthur和Jobin[18]研究发现,肠道某些细菌可以通过产生毒素引起结肠炎进一步诱发癌变。Rubinstein等[19]研究结果发现,CRC组织分离的核梭杆菌,通过自身产生的Fad黏附素调节E钙黏蛋白可以直接诱发癌变。
来源: 中华检验医学杂志
我有话说......
Copyright © 2015-2023 杭州宇翼科技有限公司 丨 Discuz! X3.5 丨增值电信业务经营许可证:浙B2-20190572丨浙ICP备18026348号-1丨浙公网安备33010802009352号 |