|
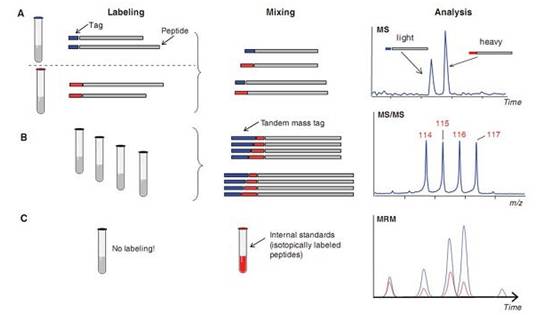
质谱仪的性能 我们已经在表1中对各种质谱仪的性能进行了一个概括。总体来说,质谱仪的性能包括分辨率、敏感度或探测极限和准确性等,这些性能都与质谱仪的类型、采用的离子化方法和扫描能力有关。不过没有哪一个仪器能同时在上述所有方面都全面占优,在选择仪器的时候必须根据实验需要进行相应的取舍。 对实验仪器性能的比较一直以来都是一个存在很多争议的话题。因为性能是服务于需求的,是取决于待测样品和实验步骤的。质谱仪对单独肽段样品进行检测时敏感度总是很低,不过如果生物样品的基质背景(matrix background)很高,那么检测的敏感度就会提高好几个数量级。这种同一款仪器在性能上表现出来的“不稳定性”其实很常见,在仪器处于不同操作条件或状态下时(比如,在最优条件下,常规条件下和大批量处理条件下)它们的性能表现都是不同的。实验目的是:想进行定量分析还是蛋白质鉴定,这也决定了该使用哪种仪器。在蛋白质鉴定试验中,仪器的分辨率(能很好地区分不同组分)和准确性是最主要的,而在定量研究中,敏感度、动态范围和MRM能力才是最主要的。因此,我们应该根据实验目的的需要以及实验设计安排来确定该使用哪种质谱仪。 要想用全扫描模式(full scanmode)获取定量数据的同时再用MS/MS模式获取定性数据是一件非常困难的事情。不过某些“杂交”质谱仪,比如LIT-ICR质谱仪,由于它们能够平行采集数据,因此可以部分解决上面那个问题。精确的定量分析需要源自整个洗脱图(entire elution profile)的高质量的、高信噪比的数据信息。数据质量与数据采集参数高度相关,这些数据采集参数包括扫描时间或者采样时间(对于非扫描质谱仪而言)。因此,我们经常需要在数据质量和样品处理能力(量)之间做出取舍。 最近又有几项有关质谱仪的最新进展问世,这些新成果的出现又给我们的生物大分子研究工作补充了“弹药”。在蛋白质测序方面,基于碰撞诱导裂解技术(CID),又新出现了可变裂解技术(Alternate fragmentation technique),该新技术是基于处在碰撞池中的离子具有的电子传递特性开发出来的。目前,电子捕获解离技术(ECD)和电子传递解离技术(ETD)都已经分别被应用到FT-ICR质谱仪和LIT质谱仪上了。运用这两种技术产生的蛋白质裂解产物能与经典的CID方法裂解产物互为补充。不过这两种新方法裂解更均匀,更适合用于发现翻译后修饰情况。ECD技术和ETD技术还都可以用于大型肽段和蛋白质的研究。他们的裂解能力和对完整蛋白的分析能力可以帮助我们直接用质谱仪对完整的蛋白质进行分析,即可以采用所谓的“自上而下(top-down)”的方法进行研究。这样,我们能够获得完整的氨基酸序列信息和翻译后修饰信息,能够对蛋白质进行最准确的鉴定。 传统的和最新的蛋白质组学研究策略 虽然到目前为止,还没有一种蛋白质组学研究策略能够对某个蛋白质组进行常规的、完整的分析,但是现在的技术已经非常强大,我们相信,很快就能进行全蛋白质组学研究了。而且,对某个亚蛋白质组(比如某个细胞器或亚细胞结构的蛋白质组)进行研究早就已经不是什么难题了,这已经成为了一种常规的研究手段。不过,蛋白质组学研究方法也需要视研究目的而做出相应的调整,不能千篇一律,比如,有很多研究都是描述性的研究项目,主要的关注点都着眼在发现、鉴定出蛋白质以及这些蛋白质的翻译后修饰情况,而最近又开始逐渐兴起蛋白质组学定量研究了。 实际上,每一个以质谱检测为基础的蛋白质组学研究工作都包括以下3大部分:(i)分离、消化蛋白质样品,然后对样品进行进一步裂解;(ii)对样品进行质谱定性和定量检测;(iii)用相应的软件对质谱检测结果进行分析处理,获得蛋白质的氨基酸序列,并且如果可能的话,进行定量分析。蛋白质的鉴定工作主要由MS-MS质谱仪负责,然后通过将质谱检测结果与数据库中的数据进行比对,最后确定出蛋白质的氨基酸序列。需要提醒的是,对结果的统计分析工作是保证结果正确性的关键。 如果给待测样品带上稳定的同位素标记,我们就可以对样品中不同蛋白质的丰度进行检测了,这些蛋白质可能在化学性质方面是一样的,但是我们可以通过不同的同位素信号强度比对它们进行区分(图3A)。也可以使用如图3B中所示的串联标记(tandem mass tag)方法对样品进行多次质谱分析。还可以在进行质谱分析之前往待测样品中添加定量的同位素标记肽段,以获得样品准确的定量数据(图3C)。 这种“鸟枪法”最大的优势就是简单,不论从理论层面来说还是从实验操作层面来说都很简单,该方法相比前面所述的各种方法能够对蛋白质组分进行更大程度的覆盖,同时定量的准确性更高。不过该方法也有其局限性,具体表现在动态范围不够,难以使用生物信息学方法从大量的、高冗余的、复杂的蛋白质组样品质谱数据中推断出蛋白质序列。幸好这些问题已经部分得到了解决,通过裂解的方法可以降低样品的复杂程度。常用的裂解方法主要针对的都是蛋白质组中生物信息学资料丰富的部分,比如富含半胱氨酸的肽段、磷酸化的肽段、糖基化的肽段等等。这种“鸟枪法”策略最适合用于快速鉴定复杂样品中的组分,也非常适合用于对不同样品中同一蛋白质的定量比较研究。在“鸟枪法”策略中,在蛋白质水解过程里,不同肽段间的联系信息以及肽段与其来源蛋白质间的联系信息都已经丢失了,因此该方法不太适合用于对具有多重修饰的蛋白质进行鉴定工作。
Protein(s):待测蛋白质样品;Enz.Digestion:酶解;Pep. Mixture:裂解产物混合物; MS Analysis:质谱检测分析;DB Search:数据库比对搜索;Identities:鉴定;
A:经由2维电泳或pull-down实验发现的待测蛋白质样品prot随即被酶解,然后用质谱仪对酶解片段pep进行质谱检测分析。最后根据肽质指纹图谱(PMF)鉴定出肽段以及它们的来源蛋白。其它的MS/MS数据也能用来进行肽段鉴定。 B:随机蛋白质鉴定和定量分析方法,即鸟枪法。该方法可以对样品同时进行鉴定以及定量研究。被选出的肽段如图1A中所示,经过串联质谱仪进行离子扫描分析。在这里,母离子是被随机选出的,通常母离子的裂解片段中只有一个片段能够被检测到,也就是说存在采样不足的问题。MS1质谱仪采集到的离子强度信息与参考分子信息比对可以进行定量分析,这里使用的参考分子通常都会标记上稳定的同位素标签。 C:定量研究方法能去除蛋白质定量研究与鉴定过程中的干扰信号。首先,MS1质谱仪会通过比对不同样品的肽模式,即肽段分子量相对色谱保留时间作图的结果,挑选出不同样品间丰度不同的肽段。然后被挑出的肽段进入下一轮MS/MS质谱检测。 D:基于各种假说的检测方法。该方法可以对各种预先被设定肽段的丰度进行高精度的检测,我们通常都是根据以往的实验结果来挑选这些靶肽段。在这里常用的方法是图1D中所示的MRM方法。如果在试验中选用合适的参考肽段会进一步提高实验的准确性。
比较模式分析 基于各种假说的研究策略 上述这种研究策略最适合用于Q-Q-LIT质谱仪这类被我们使用了几十年,主要用于检测小分子药品和体内药物代谢研究领域的三重四级杆质谱仪。在研究药物代谢时使用的研究方法同样适用于蛋白质组学研究领域。 作为上述方法的一个补充,Smith又开发出另一种使用精确分子量标签来鉴定蛋白质的方法。该方法首先测定待测物质的分子量,然后将数据与分子量数据库进行比对,以此来鉴定蛋白质。这样就无需再对每一个样品中的每一条多肽进行测序了。随着蛋白质组学研究的不断开展,我们积累的数据也越来越多,并且数据积累的速度也越来越快,同时,各种分析软件的功能也越来越强大,因此我们相信,基于各种假说的蛋白质组学研究策略一定会被更多的人所接受,所采用。
Labeling:标记 Tag:标记物 Peptide:肽段 Mixing:混合 Analysis:分析 图3 肽段定量分析策略示意图。 A:同位素稀释法是进行肽段定量分析时最常用的一种方法,该方法也是进行蛋白质组学研究时常用的一个方法。该方法的原理是在一号样品所有的待测蛋白质上都带上一个稳定的同位素标签,同时在二号样品所有的待测蛋白质上都带上另一个稳定的同位素标签。这样,这两组样品就可以互为参照了。可以借助化学的方法,例如稳定的同位素编码标签试剂、代谢标记反应和酶标反应等对蛋白质进行同位素标记。 B:使用串联标签进行定量研究。该方法同样需要各种稳定的同位素标记物。这些同位素标记物由两种同位素标记元素组成,它们都有固定的分子量。目前,这些试剂可用于四通道反应。通过在质谱仪的MS/MS模式下检测连接在蛋白质N端报告基团的相对强度以及CID图谱中低分子量范围里的信号可以获得待测蛋白质的定量信息。 C:使用内参进行定量研究的方法。该方法实际上也是一种同位素稀释法。在该方法中会往待测样品中添加一种已知浓度的同位素标记的肽段,然后借助标准曲线进行精确的定量分析。虽然该方法样品制备过程较为复杂,但是它的前景非常光明。它非常适合用于基于各种假说的检测方法。 在过去的十年里,是蛋白质分析,更准确的说应该是蛋白质组学分析推动了质谱检测技术的发展。各种技术进步已经使质谱仪在准确度、分辨力、敏感度、定量分析能力等各方面都有了长足的进展,蛋白质组质谱检测策略方面也有了新突破。分析完整蛋白质、蛋白质复合体、低丰度蛋白质等各种样品的检测操作流程层出不穷。虽然这些质谱检测技术是为了满足蛋白质分析的需求而诞生的,但是它们出现之后又反过来推动了生物大分子(包括代谢产物、脂类物质、碳水化合物等等)领域的研究。因此,我们有理由相信,质谱检测技术必将在生物研究的各个领域里占有重要的一席之地。 原文检索: |
Copyright © 2015-2023 杭州宇翼科技有限公司 丨 Discuz! X3.5 丨增值电信业务经营许可证:浙B2-20190572丨浙ICP备18026348号-1丨浙公网安备33010802009352号