|
《CAP TODAY 》和分子病理学协会已经合作,将分子病例报告带给《CAP TODAY 》读者。AMP成员使用他们自己的实践中的临床案例撰写报告,这些案例显示了分子测试在诊断、预后和治疗中的重要作用。以下报告来自德克萨斯大学西南医学中心。
Jing Xu, MD, PhD; Guanglu Shi, PhDOzlem Kulak, MD, PhD; Weina Chen, MD, PhDPrasad Koduru, PhD; Jeffrey Gagan, MD, PhD慢性淋巴细胞白血病/小淋巴细胞淋巴瘤(CLL / SLL)是最常见的淋巴增生性疾病之一。它是CD5阳性的B细胞肿瘤。CLL / SLL的特征之一是它的异质性,不仅在个体之间,而且在个体患者中也是如此。1细胞遗传学和分子变异在疾病发展过程中以及对靶向疗法的反应中是动态的。在这里,我们将介绍一名CLL / SLL患者,其疾病在疾病进展的不同阶段以分子和细胞遗传学进化为特征,最终发展到弥漫性大B细胞淋巴瘤(Richter综合征)。案例:尽管进行了相应的治疗,但一名53岁的男性于2019年就诊于我院,仍患有CLL / SLL进行性疾病。该患者于2004年首次在一家医院通过淋巴结活检诊断为CLL / SLL,原因是他的颈部淋巴结肿大。流式细胞仪对CD38和ZAP-70呈阳性。当时没有进行细胞遗传学或分子测试。然后,他接受了氟达拉滨/利妥昔单抗的治疗,反应良好。他的疾病发展的第一个实例发生在2007年,当时他接受了氟达拉滨/环磷酰胺/利妥昔单抗的治疗,并再次表现出良好的反应。下一次在2010年观察到进行性淋巴结肿大。当时的骨髓活检显示CLL / SLL广泛侵犯了骨髓。染色体核型和CLL / SLL FISH面板未显示任何异常。然后用苯达莫司汀/利妥昔单抗治疗他,仍然无法控制这种疾病。2012年的另一次骨髓活检显示,有44%的CLL / SLL细胞累及骨髓。核型分析显示13q缺失。然后,他接受了依鲁替尼和利妥昔单抗治疗四年。2016年,他再次经历了进行性淋巴结病。在一家外部医院对他的骨髓样本进行了下一代测序测定。它涵盖了29个单核苷酸变体的基因,以及插入和缺失(indels)。它显示了BTK中的突变,与依鲁替尼(Ibrutinib)耐药性相关,以及SF3B1和XPO1基因的突变(表1)。顺式中的两个TP53点突变尽管没有报道确切的等位基因频率,但也检测到了比其他变体更低的等位基因频率。因此,他从依鲁替尼转为使用维奈妥拉 /利妥昔单抗,后来又转为依达拉西布/利妥昔单抗治疗。这些疗法均未成功控制该疾病。2019年2月,PET CT显示腹腔内腺病伴明显肝脾肿大。腹膜后病变的图像引导活检显示CLL / SLL的淋巴细胞转化,没有大细胞转化的迹象。然后,他于2019年3月接受CD19 CAR T细胞疗法。通过流式细胞术分析,CAR T细胞疗法后的骨髓活检显示MRD阴性。但是,一个月后进行的PET扫描显示代谢亢进性腺病。因此,重新开始使用依鲁替尼治疗。2019年11月在我们医院进行的再次骨髓活检显示CD5(+)/ CD10(-)弥漫性大B细胞淋巴瘤浸润,可能代表Richter综合征(图1)。淋巴瘤细胞通过免疫组织化学对MYC,BCL2,MUM-1和TP53呈阳性,但对BCL6呈阴性。骨髓抽吸物的核型异常,其中三个相关克隆含有多个结构异常,包括重排导致额外复制的8q和11q和17p缺失(图2)。与染色体核型结果一致,FISH研究还显示出MYC(8q24)(在被检细胞的92.5%至96.5%中)和ATM(11q22.3)(在被检细胞的91.5%中)的额外复制和TP53(17p13.1)缺失的证据(在80%的检测细胞中)(图3)。值得注意的是,通过核型或FISH分析未在该骨髓中检测到2012年样品中见到的del(13q)异常。免疫球蛋白重链变量(IGHV)基因未突变,与Richter综合征和较差的临床预后相关。2,3
A)散在的大淋巴瘤细胞(红色箭头)在骨髓穿刺液中有明显的核仁(BMA,Wright-Giemsa染色,×500);B)在BM凝块切片中的大淋巴瘤细胞片(H&E染色,×500);C和D)淋巴瘤细胞在C中表达B细胞标记(PAX5),在D中表达TP53(免疫组化染色,×200)。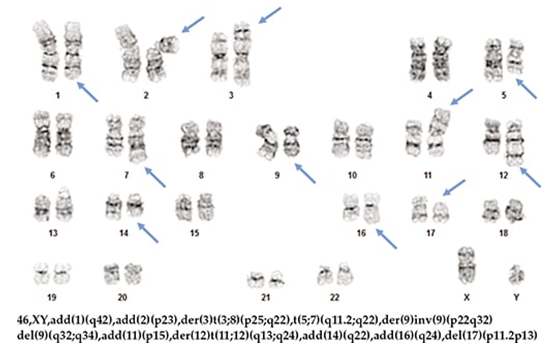
图2.在2019年骨髓样本中鉴定出的克隆之一的核型两种NGS测试均检测到SF3B1和TP53基因的点突变,尽管它们的密码子不同,这表明这些突变可能代表了仅由XPO1突变的祖先的亚克隆突变。实际上,据报道SF3B1和TP53突变更常见于亚克隆,并且代表CLL / SLL中的晚期事件。5个两者TP53和SF3B1突变与里氏综合征和预后不良的风险增加相关联。12,13
图3. FISH测试显示MYC (A),ATM (B)和TP53 (C)缺失的额外副本。没有检测到RB1缺失的证据(D)。为了进一步表征该疾病,我们使用我们机构的1,385个基因组(完整基因列表位于:https://j.mp/3oYr2bf)对外周血样本进行了NGS研究。该测定法报告了单核苷酸变体,插入缺失,拷贝数和基因融合。检测到多个基因的突变,包括TP53,XPO1,SF3B1,BTG1和KRAS(表1)。有趣的是,只有XPO1突变与2016年样本一致。但是,2016年的检测没有询问BTG1或KRAS。尽管TP53和SF3B1在两个样品中都发生了突变,但观察到的变异却完全不同。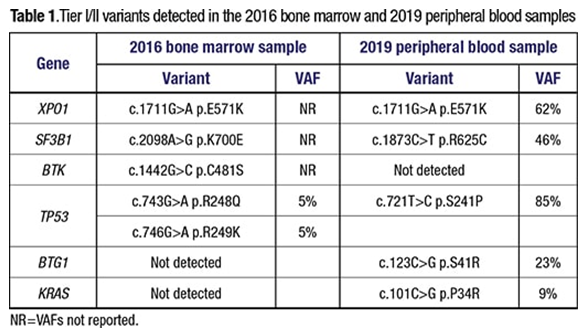
患者进一步接受了临床试验ARQ531,然后接受了DA-R-EPOCH /依鲁替尼治疗。不幸的是,尽管进行了这些治疗,该疾病仍在继续发展,并且患者由于肺部并发症而离世。没有进行尸检。讨论:CLL / SLL的特征是其病程的高度可变性。虽然有些患者病情恶化,但另一些则发展为难治性疾病,需要进行化学疗法。在大约5%至10%的患者中,CLL / SLL可以转变为侵袭性淋巴瘤,最常见的是DLBCL。4 CLL / SLL的进展是一个复杂而动态的过程,涉及不同亚克隆的发展,其优势随着时间的推移而变化。据报道,驱动突变可分为两类:克隆突变,存在于所有白血病细胞中,代表了肿瘤发生过程中的早期事件;亚克隆突变,它们存在于肿瘤细胞的一个子集中,代表晚期事件。5亚克隆突变的数量通常随着治疗而增加。5在这种情况下,在疾病的不同时间点(2010年,2012年,2019年)进行了三项细胞遗传学测试,结果不同。虽然第一次测试正常,但是2012年进行的第二次测试在白血病细胞中缺失了13q,而2019年进行的第三次测试则显示了三个相关克隆的复杂核型,这些克隆具有多个结构异常,包括额外的8q和11q复制,以及17p缺失(表2)。13q缺失是CLL / SLL中观察到的最常见的染色体异常之一,如果是唯一的遗传异常,则与良好的预后相关。6复杂的核型和17p缺失与Richter综合征和不良预后相关。3,6这三种细胞遗传学结果的差异与白血病细胞的克隆和亚克隆进化理论一致。此外,在2012年的样品中未发现2012年样品中检测到的13q缺失异常这一事实增加了以下可能性:这些是最早可追溯至2012年的肿瘤细胞的不同亚群。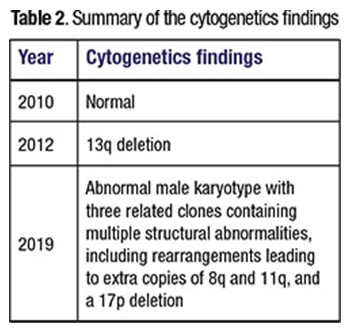
在2016年和2019年进行的两次NGS测试也获得了与众不同的结果,这引发了一个疑问,即DLBCL是否实际上代表了新生肿瘤。该XPO1 p.E571K突变是在两个测试中始终检测到的唯一变量。XPO1(exportin)是核转运蛋白importin-β超家族的成员,它介导许多蛋白质和RNA向细胞质的转运,从而调节关键的信号传导途径和细胞功能。E571K是一个热点突变,已显示出它会改变XPO1对核输出信号的亲和力。7值得注意的是,据报道XPO1突变与Richter综合征8-10有关,但在从头DLBCL 11中很少发现这支持了DLBCL来自先前的CLL / SLL的观点。该XPO1 E571K突变可能代表的克隆突变。亚克隆进化和遗传分化随后开始,并导致细胞具有不同的突变和染色体异常。我们的患者病程较长,经过多次治疗和复发。在疾病进展过程中也鉴定出该患者的耐药性突变。2016年采集的患者骨髓样本中最初检测到BTK p.C481S突变,当时他发展为依鲁替尼治疗难治性疾病。C481S是特征明确的依鲁替尼耐药突变,可破坏依鲁替尼与BTK之间不可逆的共价结合。14在2019年的样本中未检测到此突变,这表明具有耐药性突变的亚克隆可能会在没有药物选择压力且不会屈服于其他亚克隆的情况下失去优势。一个BTG1在他的维奈妥拉 /利妥昔单抗,艾达拉西布/ 利妥昔单抗和CAR T细胞疗法失败后,于2019年外周血样本中检测到p.S41R突变。BTG1是调节细胞生长和分化的抗增殖基因家族的成员。值得注意的是,BTG1突变未处理的CLL / SLL很少检测到,但已经与抗维奈妥拉有关。15总之,CLL / SLL是高度可变的疾病,其特征在于白血病细胞的克隆和亚克隆进化。对应于治疗选择,新的突变可能出现或消失。因此,使用NGS调整个体治疗方案来纵向监测分子分布是有益的,尤其是在发生疾病转化时。1.Guièze R, Wu CJ. Genomic and epigenomic heterogeneity in chronic lymphocytic leukemia. Blood. 2015;126(4):445–453.2.Hallek M, Cheson BD, Catovsky D, et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood. 2018;131(25):2745–2760.3.Niederhuber JE, Armitage JO, Doroshow JH, Kastan MB, Tepper JE. Abeloff’s Clinical Oncology. 6th ed. Elsevier; 2020.4.Parikh SA, Shanafelt TD. Risk factors for Richter syndrome in chronic lymphocytic leukemia. Curr Hematol Malig Rep. 2014;9(3):294–299.5.Landau DA, Carter SL, Stojanov P, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell. 2013;152(4):714–726.6.Dierlamm J, Michaux L, Criel A, Wlodarska I, Van den Berghe H, Hossfeld DK. Genetic abnormalities in chronic lymphocytic leukemia and their clinical and prognostic implications. Cancer Genet Cytogenet. 1997;94(1):27–35.7.García-Santisteban I, Arregi I, Alonso-Mariño M, et al. A cellular reporter to evaluate CRM1 nuclear export activity: functional analysis of the cancer-related mutant E571K. Cell Mol Life Sci. 2016;73(24):4685–4699.8.Walker JS, Hing ZA, Harrington B, et al. Recurrent XPO1 mutations identified as predictive marker for advanced CLL progression. HemaSphere. 2019;3(Suppl 1):508.9.Eyre TA, Schuh A. An update for Richter syndrome—new directions and developments. Br J Haematol. 2017;178(4):508–520.10.Stamatopoulos B, Antoniou P, Vavoulis D, et al. Characterization of recurrent mutations in patient with a Richter syndrome by targeted next generation sequencing. Blood. 2016;128(22):3200.11.Jardin F, Pujals A, Pelletier L, et al. Recurrent mutations of the exportin 1 gene (XPO1) and their impact on selective inhibitor of nuclear export compounds sensitivity in primary mediastinal B-cell lymphoma. Am J Hematol. 2016;91(9):923–930.12.Nadeu F, Delgado J, Royo C, et al. Clinical impact of clonal and subclonal TP53, SF3B1, BIRC3, NOTCH1, and ATM mutations in chronic lymphocytic leukemia. Blood. 2016;127(17):2122–2130.13.Fabbri G, Khiabanian H, Holmes AB, et al. Genetic lesions associated with chronic lymphocytic leukemia transformation to Richter syndrome. J Exp Med. 2013;210(11):2273–2288.14.Woyach JA, Furman RR, Liu TM, et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med. 2014;370(24):2286–2294.15.Herling CD, Abedpour N, Weiss J, et al. Clonal dynamics towards the development of venetoclax resistance in chronic lymphocytic leukemia. Nat Commun. 2018;9(1):727.文章来源于:https://www.captodayonline.com/ |



