|
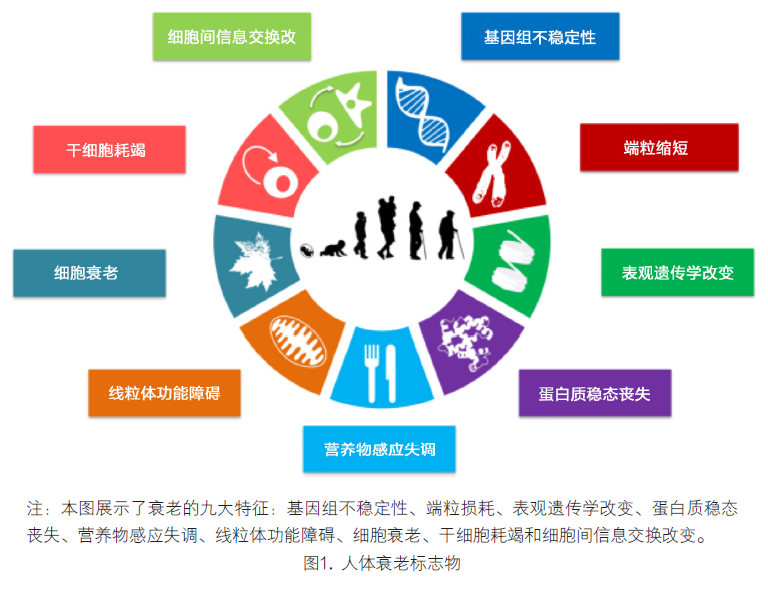
广义地讲,衰老可被定义为大多数生物体均可发生的增龄性功能减退。在人类历史进程中衰老总是会激发人们的好奇和联想。30多年前(1983年),对秀丽隐杆线虫首个长寿品系的分离(Klass, 1983),开启了衰老研究的新纪元。如今,基于对生命和疾病的分子及细胞基础的全面认识,衰老现象正经受着全面的科学审视。当前的衰老研究和数十年以来肿瘤方面的研究有着诸多相似之处。2000年一篇里程碑式的文献使肿瘤研究领域获得了强劲的动力,该文献归纳了肿瘤的六项特征(Hanahan and Weinberg, 2000),近期扩展为十项(Hanahan and Weinberg, 2011)。通过归纳,概念化地呈现了肿瘤的本质及其深层机制。 初看肿瘤和衰老似乎是对立的过程:肿瘤是细胞适应性异常的结果,而衰老则以细胞适应性丧失为特征。而深入来看肿瘤和衰老具有某些共同的根源。细胞损伤的增龄性蓄积是普遍认为的衰老原因(Gems and Partridge, 2013; Kirkwood, 2005; Vijg and Campisi,2008),而细胞损伤又为特定细胞的癌变提供有利条件,并最终导致肿瘤的发生。因此,肿瘤和衰老可被看作是同一基础过程,即细胞损伤蓄积的两种不同表现。另外,诸如动脉粥样硬化、炎症等衰老相关病变,还涉及到细胞失控性过度生长或功能活跃(Blagosklonny,2008)。基于上述概念框架,衰老领域应关注一系列关于衰老损伤生理性机制的关键性问题;如试图重建体内稳态的代偿性反应,不同损伤和代偿反应之间的相互联系,外源性干预延缓衰老的可能性。 本文试图对衰老的细胞和分子特征予以确认和归类。本文提出的九项特征已被普遍认为能够促进衰老的进展,并可共同决定衰老的表型(图1)。考虑到衰老特征的复杂性,本文重点关注针对哺乳动物衰老的最新认识,同时也会对低等模式生物的相关研究有所提及(Gems and Partridge,2013;Kenyon,2010)。各项衰老特征应严格符合以下标准:(1)其应在正常衰老过程中显现;(2)实验对其增强后应加速衰老;(3)实验对其削弱后应能够延缓正常衰老进程并由此增加健康寿命。本文提出的九项特征均不同程度的符合这套严格的标准,具体内容将在下文一一讨论。这套标准的最后一条是最难实现的,即使将其局限于衰老的某一方面亦是如此。因此,九项特征不是全部满足“通过干预成功延缓衰老”这一条件。上述各项衰老特征之间存在广泛关联,这意味着通过实验改变某一特征,可能会影响到其它特征。
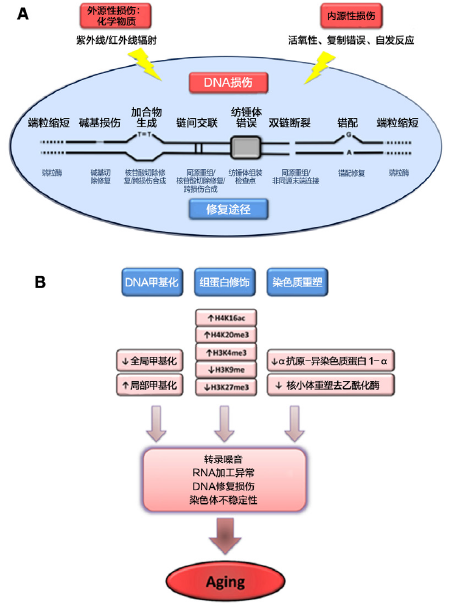
基因组不稳定性 衰老的一个共同点是贯穿生命过程中的基因损伤累积(Moskalev et al., 2012)(图2A)。多种早老性疾病,如Werner综合征和Bloom综合征,均由DNA损伤增加所致(Burtner and Kennedy,2010),但是上述疾病及其他早老综合征与正常衰老之间的相关性尚未阐明,部分原因在于这些疾病仅能概括衰老的某些方面。外源性因素(如物理、化学和生物因子)和内源性因素,如DNA复制错误、自发性分解反应、活性氧(ROS)(Hoeijmakers,2009)等均可破坏DNA的完整性和稳定性。外源性或内源性损害造成的基因损伤类型各异,包括可由各种病毒或转座子共同作用所导致的点突变、易位、染色体获得或缺失、端粒缩短和基因断裂等。为尽可能减少上述损伤,机体DNA修复机制进化形成一个复杂网络,能够协同对抗针对细胞核DNA的大多数损害(Lord and Ashworth,2012)。基因组稳定系统还包括某些特殊机制,能够使端粒保持适当长度和功能(端粒与衰老的另一特征有关,见下文)以及确保线粒体DNA的完整性(Blackburn et al., 2006;Kazak et al., 2012)。除外DNA的直接损伤,细胞核结构的缺陷,即核纤层蛋白病,亦可引发基因组不稳定性并导致早老综合征(Worman,2012)。 1 . 细胞核DNA:老年人和老年模式生物的细胞均会表现出体细胞突变的累积(Moskalev et al.,2012)。而其他类型的DNA损伤,如染色体非整倍体和拷贝数变异等现象亦被发现与衰老相关(Faggioli et al., 2012;Forsberg et al., 2012)。还有报道,大型染色体异常中的克隆镶嵌现象亦有所增加(Jacobs et al., 2012;Laurie et al., 2012)。上述各种类型的DNA改变均可影响到基本基因及其转录途径而导致细胞功能紊乱,若未通过凋亡或衰老而被清除,则会危及组织和机体的稳态。尤其当DNA损伤影响到干细胞的特定功能时,则会对组织再生造成影响(Jones and Rando,2011;Rossi et al., 2008)(见干细胞耗竭部分)。小鼠和人的研究发现生命过程中基因组损伤的增加与衰老呈现因果关系,表明DNA修复机制的缺陷会导致小鼠衰老加速,以及人类的多种早老综合征的发病,如Werner综合征、Bloom综合征、着色性干皮病、毛发硫营养不良、Cockayne综合征、Seckel综合征等(Gregg et al.,2012;Hoeijmakers,2009;Murga et al., 2009)。另外,在转基因小鼠中过表达BubR1(一种确保染色体精确分离的有丝分裂检查点组分),可增强对抗染色体非整倍性和恶性肿瘤的防御能力,并延长健康寿命(Baker et al., 2013)。这些发现提供了通过人为加强核DNA修复机制可以延缓衰老进程的实验证据。 2. 线粒体DNA:线粒体DNA(mtDNA)的突变和缺失也可促进衰老(Park and Larsson,2011),mtDNA被认为是衰老相关体细胞突变的主要靶点。原因在于线粒体的氧化微环境,mtDNA缺乏组蛋白的保护,而且与核DNA相比,mtDNA修复效率低下(Linnane et al.,1989)。由于线粒体基因组具有多重性,同一细胞中可并存突变型和野生型基因组,这一现象称为“异质性”。关于衰老mtDNA突变的因果关系的推测尚存争议。单细胞分析显示,尽管mtDNA突变的总体水平较低,但个体的衰老细胞突变负荷较显著,形成以突变基因组为主并达到同质性状态(Khrapko et al., 1999)。有趣的是与早期的预测相反,成年或老年细胞中多数mtDNA突变是由生命早期的复制错误所致,而非氧化损伤。这些突变会发生多克隆增殖,导致不同组织出现呼吸链功能障碍(Ameur et al., 2011)。HIV感染者经抗逆转录病毒药物(可干扰mtDNA复制)治疗后,会出现衰老加速,这支持了生命早期mtDNA突变的多克隆增殖加速衰老的观点(Payne et al., 2011)。 mtDNA损伤与衰老及增龄性疾病相关的最早证据来自mtDNA突变所致人类多系统疾病可模拟某些衰老表型(Wallace,2005)。更深入的因果关系证据来自于对mtDNA聚合酶γ缺陷小鼠的研究。这种突变小鼠有多种早老表现且寿命缩短,这与mtDNA随机点突变和缺失的累积有关(Kujoth et al., 2005;Trifunovic et al.,2004;Vermulst et al., 2008)。来源于此小鼠的细胞表现为线粒体功能受损,但出乎意料的是,并没有伴有ROS生成增加(Edgar et al., 2009;Hiona et al.,2010)。此外,这种早老症小鼠的干细胞对mtDNA突变的累积异常敏感(Ahlqvist et al., 2012)(见干细胞耗竭部分)。需要深入研究的是能否通过基因操作降低mtDNA突变而延长寿命。 3. 细胞核结构:除了涉及到细胞核及线粒体DNA的基因损伤外,核纤层缺陷也会导致基因组不稳定(Dechat et al., 2008)。核纤层蛋白是核纤层的主要成分,且可通过充当“脚手架”以牵曳染色质和蛋白复合物参与到基因组维护中(Gonzalez-Suarez et al.,2009;Liu et al., 2005)。核纤层中编码蛋白组分的基因突变及影响其成熟和动力学的因子可导致衰老综合征如Hutchinson-Gilford早老综合征和Néstor-Guillermo早老综合征((HGPS 和NGPS)(Cabanillas et al.,2011;De Sandre-Giovannoli et al., 2003;Eriksson et al., 2003)),这一发现引起了衰老研究人员对核纤层的注意。在人类正常衰老过程中,亦可发现核纤层改变和早老素(突变核纤层蛋白前体A亚型)生成(Ragnauth et al., 2010;Scaffidi and Misteli,2006)。正常人成纤维细胞经体外长期培养,其端粒功能障碍亦可促进早老素生成。这提示在正常衰老过程中,端粒维护与早老素表达之间存在着密切关联(Cao et al., 2011)。不仅存在核纤层蛋白A的增龄性改变,核纤层蛋白B1水平亦随细胞衰老而降低,提示其可作为衰老过程中的生物标志物(Freund et al., 2012;Shimi et al., 2011)。 通过动物和细胞模型,已明确HGPS(早老症)特征性的核纤层异常可激活多条应激通路。这些通路包括p53激活(Varela et al., 2005)、生长轴失调(Mariño et al., 2010)以及成体干细胞损耗(Espada et al., 2008;Scaffidi and Misteli,2008)。观察发现,降低HGPS模型小鼠核纤层蛋白前体A或早老蛋白水平,可延缓早老症状的出现并延长寿命,这支持了核纤层异常与过早衰老之间的因果关系。而通过系统性注射反义寡核苷酸、法尼基转移酶抑制剂,或合用他汀类与氨基双膦酸盐(Osorio et al., 2011;Varela et al., 2008;Yang et al., 2006),也可取得上述效果。通过激素治疗恢复生长轴功能,或抑制NF-κB信号通路,亦可延长早老小鼠的寿命(Mariño et al., 2010; Osorio et al.,2012)。此外,对取自HGPS患者的诱导多能干细胞,采用同源重组策略可纠正其核纤层蛋白突变,这为今后开展细胞疗法开辟了道路(Liu et al., 2011b)。而能否通过强化核结构来延缓正常衰老,尚有待在今后研究中予以验证。 4. 小结:大量证据显示衰老过程伴随着基因组损伤,而人为诱导基因组损伤则会导致某些衰老加速。机体存在确保染色体准确分离的机制,遗传学证据显示,加强这种机制能够延长哺乳动物寿命(Baker et al., 2013)。一些特殊的早老症与核结构缺陷相关,而相关治疗可延缓此类早老症。寻找干预措施以加强细胞核和线粒体基因组的稳定性(如DNA修复)以及它们对正常衰老的影响,也是需要研究的方向(端粒问题较为特殊,以下单独讨论)。 端粒缩短 DNA损伤的增龄性累积对基因组的影响似乎是随机的,但在染色体的某些区域(如端粒)则特别容易发生增龄性损害(Blackburn et al., 2006)(图2A)。复制性DNA聚合酶不具备完全复制线性DNA分子末端的能力,而一种特异的DNA聚合酶(即端粒酶)则具备这种功能。然而,多数哺乳动物的体细胞不表达端粒酶,这导致了染色体末端端粒保护序列进行性和累积性的丧失。端粒缩短解释了某些类型的离体培养细胞增殖能力有限的原因,这种现象称为复制性衰老,或称为Hayflick极限(Hayflick and Moorhead,1961;Olovnikov, 1996)。事实上,通过异位表达端粒酶可有效实现普通细胞永生化,且不会发生致癌性转化(Bodnar et al., 1998)。重要的是端粒缩短在人类和小鼠的正常衰老过程中也可发现(Blasco,2007a)。 端粒缩短可以被视为躲避了DNA修复机制的DNA断裂(Palm and de Lange,2008)。端粒通过形成具有特异性保护作用的端粒蛋白复合体躲避DNA修复机制。端粒的这一特殊性使端粒不仅在没有端粒酶的情况下逐渐缩短,即使在端粒酶存在的情况下,由于端粒蛋白复合体的存在,外源性DNA损伤造成的端粒破坏也会逃避DNA修复机制。因此,端粒产生持久的DNA损伤会导致包括衰老和/或凋亡在内的有害的细胞效应(Fumagalli et al., 2012;Hewitt et al.,2012)。 人类端粒酶缺陷与某些疾病的过早发生相关,如肺纤维化、先天性角化不良、再生障碍性贫血等,这些疾病均涉及到不同组织再生能力的丧失(Armanios and Blackburn,2012)。而端粒蛋白复合体组分的缺陷也会导致严重的端粒脱帽现象(Palm and de Lange,2008)。端粒特异性保护蛋白复合物突变在某些再生障碍性贫血和先天性角化不良病例中亦有发现(Savage et al., 2008;Walne et al., 2008;Zhong et al., 2011)。多项模型研究显示,若端粒蛋白复合体各组分功能丧失,其组织再生能力下降且衰老加速,即使端粒仍处于正常长度这一现象依然存在(Martínez and Blasco,2010)。 通过基因修饰动物模型可在端粒丧失与细胞衰老、 机体衰老之间建立因果联系。端粒缩短或延长的小鼠分别表现为寿命缩短或延长(Armanios et al., 2009;Blasco et al., 1997;Herrera et al., 1999;Rudolph et al., 1999;Tomás-Loba et al., 2008)。近期证据亦显示,通过激活端粒酶可逆转衰老,特别是针对端粒酶缺陷的小鼠。在其老年阶段采用基因手段重新激活端粒酶,则该小鼠的早老症状能够逆转(Jaskelioff et al., 2011)。此外,通过对成年野生型小鼠端粒酶采用药理激活或系统性病毒转导可延缓其正常生理性衰老,且肿瘤发病率未见增加(Bernardes de Jesus et al., 2012)。近期荟萃分析结果亦表明人类端粒缩短与死亡风险有强相关性,且在较年轻个体尤为显著(Boonekamp et al., 2013)。 小结:哺乳动物的正常衰老过程伴随着端粒缩短。并且,病理性端粒功能障碍会加速小鼠和人类的衰老。通过实验性刺激端粒酶则能延缓小鼠衰老。因此,端粒缩短完全符合衰老特征的认定标准。 表观遗传改变 各种表观遗传学改变会终生影响到所有的细胞和组织((Talens et al., 2012)(图2B)。表观遗传学改变包括DNA甲基化模式改变、组蛋白翻译后修饰以及染色质重塑。而H4K16乙酰化、H4K20三甲基化和H3K4三甲基化程度增加,以及H3K9甲基化、H3K27三甲基化程度降低,构成了表观遗传学的增龄性标志(Fraga and Esteller,2007;Han and Brunet,2012)。多种酶类系统能够确保表观遗传模式的形成和维护,如DNA甲基转移酶、组蛋白乙酰化酶、去乙酰化酶、甲基化酶、去甲基化酶以及染色体重塑相关蛋白复合体。
1. 组蛋白修饰:在无脊椎动物中,组蛋白甲基化符合衰老特征的认定标准。组蛋白甲基化复合物组分的缺失可延长线虫和果蝇的寿命(Greer et al., 2010;Siebold et al., 2010)。另外,组蛋白去甲基化酶通过靶向关键长寿通路,如胰岛素/胰岛素样生长因子-1信号通路来调节寿命(Jin et al., 2011)。目前尚不明确的是组蛋白修饰酶调控影响衰老的表观遗传学机制是通过影响DNA修复和基因组稳定性,还是通过影响细胞核外代谢或信号通路转录。 它与NAD依赖型去乙酰化酶和ADP核糖基转移酶相关的sirtuin(沉默信息调节因子2相关酶)家族成员已被广泛认为是潜在的抗衰老因子。人们对sirtuin蛋白家族与衰老关系的兴趣源自于一系列针对酵母、果蝇和蠕虫的研究报告。报告显示,上述生物体中唯一的sirtuin基因Sir2具有显著的长寿活性(Guarente,2011)。最初发现,酿酒酵母过表达Sir2,复制型酵母寿命会延长(Kaeberlein et al., 1999)。之后的报告提示分别过表达Sir2在蠕虫(sir-2.1)和果蝇(dSir2)的直系同源基因,亦可延长这两种无脊椎模式生物的寿命(Rogina and Helfand,2004;Tissenbaum and Guarente,2001)。不过,近期对上述研究又出现质疑,报告认为蠕虫和果蝇研究所见寿命延长,很大程度上是由于其复杂的遗传背景差异,而与过表达sir-2.1或dSir2无关(Burnett et al., 2011)。实际上,经过细致的再次评估发现,过表达sir-2.1只能中等程度延长线虫寿命(Viswanathan and Guarente,2011)。 多项研究显示,哺乳动物的7种sirtuin同源基因中有数种亦可延缓小鼠衰老的多项参数(Houtkooper et al., 2012;Sebastián et al., 2012)。具体来看,哺乳动物转基因过表达SIRT1(为最接近无脊椎动物Sir2的同源基因),可提高衰老过程中各方面的健康水平,但并不能延长寿命(Herranz et al., 2010)。然而,SIRT1良性效应的产生机制较为复杂且各机制间相互关联,包括从提高基因组稳定性(Oberdoerffer et al., 2008;Wang et al., 2008)到增强代谢效率的广泛细胞作用(Nogueiras et al., 2012)(见营养素感应失调)。而在哺乳动物中,支持sirtuin介导促长寿效应的更有力证据来自于对SIRT6的研究,其可通过组蛋白H3K9去乙酰化来调节基因组稳定性、NF-κB信号通路和血糖稳态(Kanfi et al., 2010;Kawahara et al., 2009;Zhong et al., 2010)。SIRT6缺陷的突变小鼠表现为衰老加速(Mostoslavsky et al., 2006),而过表达SIRT6的转基因雄性小鼠较对照组而言寿命更长,这与降低血清IGF-1以及IGF-1信号通路其他指标有关(Kanfi et al., 2012)。有趣的是,位于线粒体的sirtuin蛋白SIRT3亦可介导饮食限制延长寿命的某些良性效应,不过这些效应并非因为组蛋白修饰,而是因为线粒体蛋白的去乙酰化(Someya et al., 2010)。最新报道显示,衰老的造血干细胞过表达SIRT3可逆转其再生能力(Brown et al.,2013)。因此,哺乳动物sirtuin家族至少有三个成员,即SIRT1、SIRT3和SIRT6有利于健康老龄化。 2. DNA甲基化:DNA甲基化与衰老之间的关系较为复杂。早期研究描述了衰老与总体低甲基化相关,但随后的分Shumaker et al., 2006)。而所有增龄过程中出现的表观遗传学缺陷或突变会影响到干细胞的行为和功能(Pollina and Brunet,2011)(见干细胞耗竭部分)。然而,目前尚无直接实验证据显示通过改变DNA甲基化模式能够延长生物体寿命。 3. 染色质重塑:与DNA和组蛋白修饰酶相配合,染色体的关键蛋白(如异染色质蛋白1α,HP1α)和染色质重塑因子(如多梳析发现,包括对应于多种肿瘤抑制基因和多梳蛋白靶基因的多个基因座,实际上会随着增龄而高甲基化(Maegawa et al., 2010)。早老综合征患者细胞及小鼠细胞所显示的DNA甲基化和组蛋白修饰模式,在很大程度上能够概括正常衰老细胞的表现(Osorio et al., 2010;蛋白家族或NuRD复合体)的水平,在正常衰老和病理性衰老的细胞中均降低(Pegoraro et al., 2009;Pollina and Brunet,2011)。这与前述的表观遗传修饰(即组蛋白和DNA甲基化)相伴随,表观遗传因子的改变可决定染色质结构的变化(如异染色质整体缺失和再分配),这也成为了衰老的特征性表现(Oberdoerffer and Sinclair,2007;Tsurumi and Li,2012)。染色质改变与衰老的因果关系在果蝇研究中已获支持。该研究发现,HP1α功能丧失的突变果蝇寿命缩短,而过表达这种异染色质蛋白则会延长其寿命,并延缓老年期肌肉功能衰退(Larson et al., 2012)。 支持染色质表观遗传学改变与衰老之间存在功能相关性的证据还表现在DNA重复区域异染色质形成与染色体稳定性显著相关。特别是,发生在近着丝粒区的异染色质组装需要组蛋白H3K9和H4K20三甲基化,并与HP1α结合,这对染色体稳定性相当重要(Schotta et al.,2004)。对于哺乳动物,经修饰的染色质还包含丰富的端粒重复序列,这提示染色体末端可组装进入异染色质区(Blasco,2007b;Gonzalo et al., 2006)。而在亚端粒区也会显示结构性异染色质的某些特征,如H3K9和H4K20三甲基化、HP1α结合以及DNA高甲基化。因此,表观遗传学改变可直接影响到端粒长度的调节(衰老的另一特征)。此外,在DNA损伤时SIRT1和其他染色质修饰蛋白会重新定位到DNA断裂位点,从而促进修复和基因组稳定性。SIRT1除了在染色质重塑和DNA修复中的作用外,还调节蛋白质稳态,线粒体功能,营养素感应通路和炎症(见下文),这说明了衰老标志物之间存在相互联系。
来源: 国家卫健委北京医院 | 作者:王紫慧,李瑾
我有话说......
Copyright © 2015-2023 杭州宇翼科技有限公司 丨 Discuz! X3.5 丨增值电信业务经营许可证:浙B2-20190572丨浙ICP备18026348号-1丨浙公网安备33010802009352号 |