|
雄激素属于类固醇激素,主要包括睾酮(T)、雄烯二酮、脱氢表雄酮(DHEA)、硫酸脱氢表雄酮(DHEAS)、双氢睾酮(DHT)。 雄激素主要来源于肾上腺、女性的卵巢和男性睾丸的间质细胞,在类固醇激素的代谢合成途径中,雄激素作为雌激素的前体,在个体性腺分化、发育和维持生殖功能中发挥着至关重要的作用。
患者,女性,34岁,已婚,2023年3月15日于我院生殖中心门诊就诊,要求体检。
曾于2007年因无月经来潮于外院就诊,提示子宫和一侧卵巢缺如,具体不详。2017年结婚,性生活正常,未避孕、未孕。
女性外阴,阴毛稀疏,阴道通畅、呈盲端,未见明显宫颈结构,子宫区位置空虚,未扪及明显宫体,右附件未及异常,左附件区增厚无压痛,乳房有发育,乳头较小。
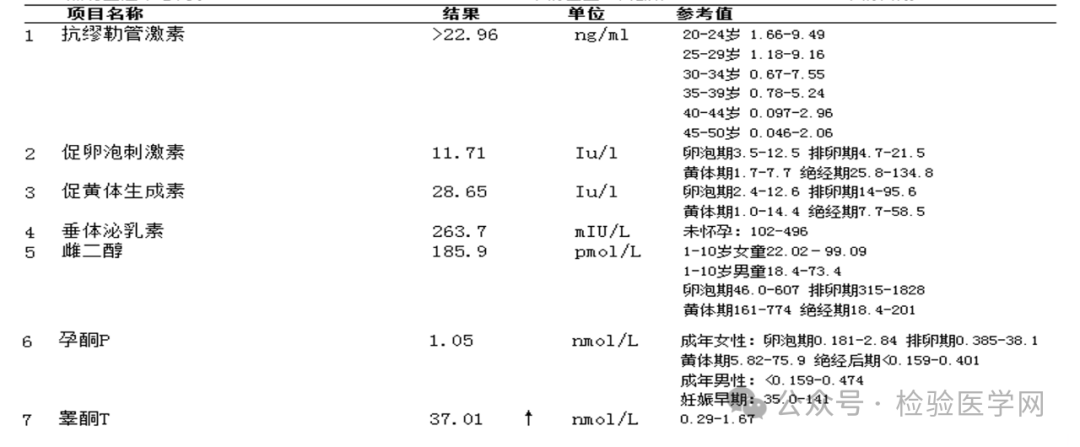
AMH >22.96ng/mL、FSH 11.71IU/L、LH 28.65IU/L、E2 185.9pmol/L、PRL 263.7mIU/L、T 37.01nmol/L、Prog 1.05nmol/L。
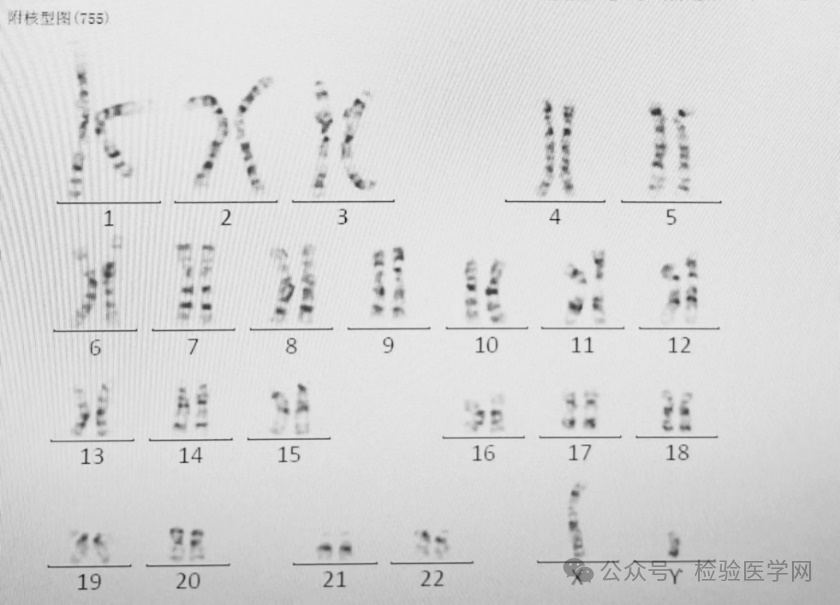
案例分析 育龄期女性睾酮主要来源于卵巢和肾上腺,各占25%,另50%由外周雄烯二酮转化而来。雄激素过高或活性增强会导致高雄激素血症,过高的雄激素可引发排卵障碍和生殖内分泌紊乱等疾病,出现月经不规律、生育力下降、多毛、痤疮、雄激素源性脱发和男性化等临床表现。睾酮升高常见于多囊卵巢综合征(PCOS)、先天性肾上腺皮质增生(CAH)、卵巢或肾上腺肿瘤等相关疾病。 本实验室性激素检测采用电化学发光法,首次检测所用平台为Cobas601,在报告单审核过程中,这例异常升高的睾酮引起了我们的注意。 为了确保结果的可靠性,我们首先就样本采集、条码粘贴、数据传输等方面分别进行核对,再查看当日两次质控情况及试剂情况,均无问题。根据我实验室复检规则:女性睾酮>3.0nmol/L时进行复检,将该样本更换至Cobas602复查,并通过倍比稀释的方式基本排除干扰和检测误差。 从正常成年女性的性激素结果来分析,该患者E2 185.9pmol/L、Prog 1.05nmol/L均为低值,表明患者目前无大卵泡,也不处于黄体期;FSH 11.7IU/L、LH 28.65IU/L均升高,LH升高更明显,但E2不高,可以基本排除排卵前的峰值;LH/FSH的比值升高,T、AMH均升高,多见于PCOS的患者,但PCOS患者睾酮罕见超过4.0nmol/L,而该患者睾酮达到37.01nmol/L,甚至超过了正常成年男性睾酮的参考范围(8.64-29nmol/L),如果用PCOS来解释,是明显解释不通的。 这个异常的激素结果我们可以发吗?患者是否有肾上腺相关疾病?是否存在明显的高雄激素临床表现? 带着疑问与临床医生取得联系,通过了解得知:该患者身高约165cm,体重55kg,血压正常,肤白,面部无痤疮,颈部无黑棘皮征,乳房有发育,乳头较小,女性特征明显且无男性化表现,外阴女性外观、阴毛稀疏;无雄激素相关用药史,无肾上腺皮质增生和卵巢肿瘤等高雄激素的相关病史及临床表现。 结合妇检无子宫及阴道呈盲端的临床表现及我实验室提供的性激素结果,初步推测,患者可能是由染色体异常导致的性发育异常,并且已开具外周血染色体检查及三维超声检查。 当日下午,妇科三维超声检查所见如下:盆腔扫查可见左右两个肌性组织回声,两个肌性组织回声团在宫颈方向相连,宫颈管显示不清;左侧大小5.5×2.4×3.2cm,包膜完整,内回声欠均匀,未见明显内膜样组织回声;右侧大小5.8×1.3×2.5cm,包膜完整,内回声欠均匀,未见明显内膜样组织回声;于肌性组织回声外上方各见一囊性结构,近髂前上极水平,大小分别1.8×1.4×1.9cm(左)、2.3×1.7×2.1cm(右),规则,内透声好。
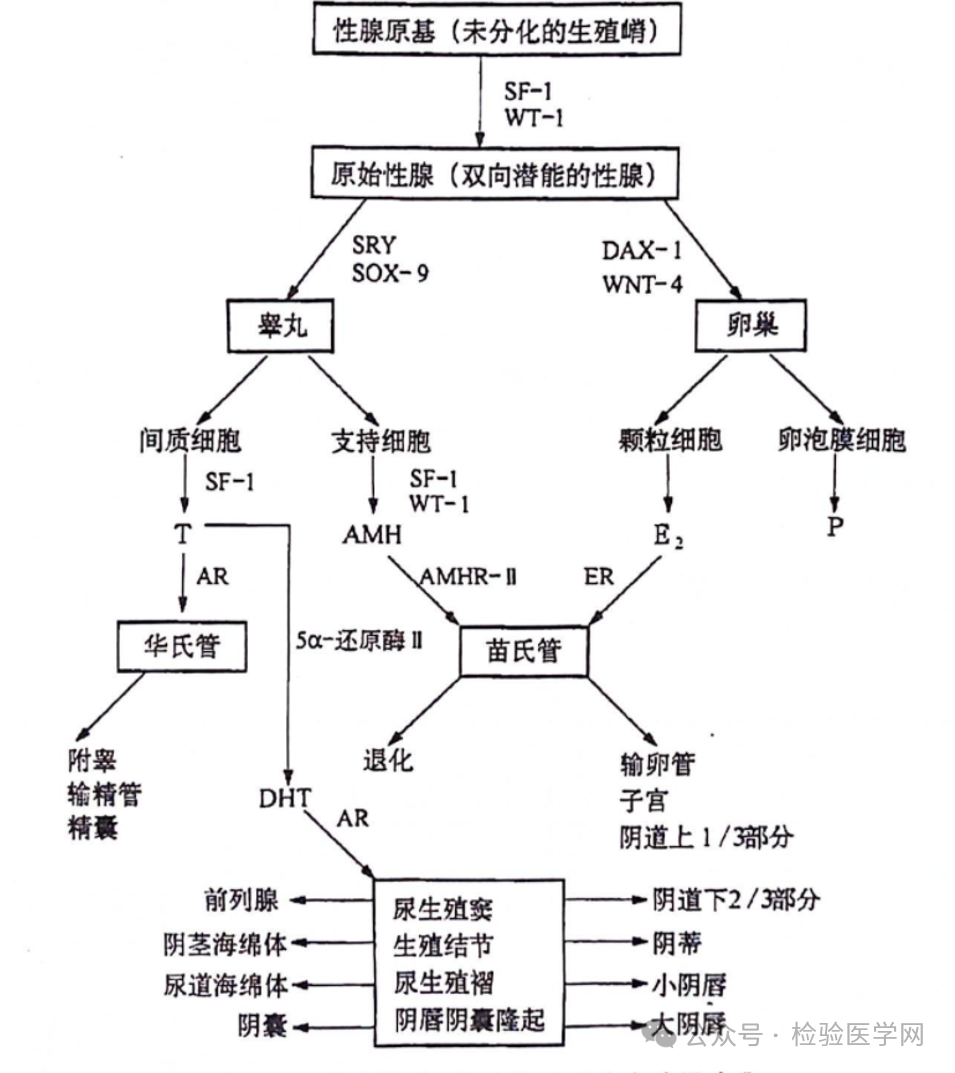
雄激素不敏感综合征(androgen insensitivity syndrome, AIS)是由雄激素受体功能障碍引起的一种常见的性发育障碍,是一种多由染色体Xq11-12上的雄激素受体(AR)突变导致的以外周雄激素抵抗为特征的X连锁隐性遗传病。根据体内雄激素受体的缺陷程度,分为完全型(CAIS)、部分型(PAIS)和轻度型(MAIS)。 CAIS是AR基因突变最严重的异常应答,患者核型为46,XY,具有女性外生殖器外形,社会性别常为女性,体内没有子宫,阴道为盲端,性腺为睾丸,无月经初潮,阴毛和腋毛的生长稀疏或缺失,在青春期可见正常的乳房发育。其发病率高达1:20000-1:60000,常见于因青春期原发性闭经就诊,在原发性闭经的病因中排第三位,占6%-10%[1]。 在正常胚胎性腺、生殖管道及外生殖器的分化和发育过程中,随着原始生殖结构的出现,原始性腺具有双向分化的潜能,有2套生殖管道,即苗氏管和华氏管。 具有Y染色体的胎儿原始性腺向睾丸方向分化,睾丸的支持细胞合成分泌抗缪勒管激素(AMH),与苗氏管的AMH II型受体结合,诱导苗氏管退化;睾丸的间质细胞合成和分泌睾酮,与雄激素受体结合,诱导华氏管发育成附睾、输精管、精囊和射精管;睾酮在5α-还原酶的作用下转化为生物学活性更强的双氢睾酮,后者与靶细胞上的雄激素受体结合,使生殖结节、尿生殖窦和生殖褶等分化形成男性外生殖器;睾酮与雄激素受体的作用进一步诱导睾丸从腹腔下降至阴囊。 在女性个体中,由于胎儿无Y染色体、无SRY决定基因,原始性腺朝卵巢分化。由于无AMH及睾酮的作用,导致华氏管逐渐退化,而卵巢产生的雌激素与苗氏管上的的雌激素受体结合,诱导苗氏管发育成输卵管、子宫及阴道的上1/3部分。由于缺乏睾酮的作用往女性外生殖器发育,尿生殖褶发育为小阴唇,生殖结节发育为阴蒂。 对于雄激素不敏感综合征的患者,染色体核型为46,XY,SRY基因使患者的性腺向睾丸方向分化,所以分泌雄激素和AMH的功能是正常的;但由于雄激素受体AR基因异常,导致雄激素不能发挥其相应的生理功能,进而影响到华氏管分化成男性内生殖器,外生殖器的发育也受阻;而AMH及其受体是正常的,AMH可使苗勒管退化,所以患者盆腔空虚,无女性的子宫及其附件。
讨论 雄激素不敏感综合征患者在新生儿和婴儿时期,实验室诊断非常困难。患者于婴儿期发病时,激素水平不太能提示雄激素抵抗,LH和T基线水平可能低下,仅在外源性人绒毛膜促性腺激素刺激之后,睾酮水平才会升[3]。 进入青春期后,由于下丘脑和垂体上雄激素受体缺陷而影响睾酮对下丘脑和垂体轴的负反馈调节作用,患者垂体分泌的LH分泌脉冲的频率和幅度均增加,FSH水平通常正常或轻度升高,升高的LH又进一步刺激睾丸间质细胞分泌更多的睾酮。在芳香化酶的作用下,雄激素可转化为雌激素,故血清睾酮水平处于典型青春期后男孩参考范围的正常高值至略微升高,雌二醇水平处于典型男性参考范围高值[4],在雌激素刺激下乳房可发育。 目前对AMH的认知大多源于卵巢功能的关注,而临床上AMH却不仅仅是针对卵巢和女性的检测。血清AMH还可用于证实睾丸功能是否正常。有研究显示,雄激素不敏感综合征患者的AMH由于接收不到睾酮的负反馈而持续升高,这与未受抑制的支持细胞功能有关[5]。我国最新发布的抗缪勒管激素临床应用专家共识(2023年版)提出,AMH结合T、抑制素B对性腺发育异常及男性不育相关疾病具有重要的应用价值[6]。 该患者为完全性雄激素不敏感综合征,根据体格检查、超声检查、内分泌激素测定和染色体核型分析不难诊断。但部分型AIS会出现部分雄激素效应,外生殖器异常呈现多样化,其诊断相对较困难,需与其他疾病引起的两性畸形相鉴别,如46,XX苗勒管发育不全(MRKH)综合征、染色体46,XY单纯性腺发育不良、17α-羟化酶缺乏症、5α-还原酶缺乏症等。
是一种女性先天性生殖道畸形,核型46,XX,有原发闭经、先天性无子宫(或始基子宫)、阴道残端等临床表现,大都可有正常的卵巢,性激素水平正常,能维持正常的雌孕激素水平,第二性征发育正常[7],不存在高雄激素血症,与本病不符。
由CYP17A1基因突变引起, 患者常于青春期以高血压、低血钾、原发性闭经、性发育异常就诊。性激素特点:FSH、LH、P水平升高,E2、T水平显著减低[8],与本病不符。
又称Swyer综合征,由SRY基因异常引起,原发闭经、第二性征不发育、外阴女性化、有子宫,激素特点:LH、FSH升高,E2、T、AMH显著减低[9]。
核型46,XY,由于缺乏5α-还原酶Ⅱ,使睾酮不能转变为二氢睾酮,睾丸分化正常,内生殖器(如附睾、精囊、输精管、射精管)正常,外生殖器表型异质性强,青春期后有第二性征发育,出现声音低沉、肌肉发达等表现[10]。 CAIS患者隐睾癌变的风险会随年龄逐渐增高,故需定期行B超、MRI等监测。一般建议在青春期后手术切除,以获得自发性乳房发育,术后激素治疗以维持第二性征[11]。对于部分性雄激素不敏感综合征的患者,可根据个人意愿和表型决定性别选择。 案例总结 完全性雄激素不敏感综合征在青春期前临床表现不明显,因此患儿及家长很少因此就医,往往进入青春期后因无月经来潮就医才被确诊,根据体格检查、激素检测、超声检查和染色体核型分析不难诊断。由于其较低的发病率和独特的临床表现,近年来也逐渐受到人们的关注。 检验人员在报告审核过程中,通过结果分析发现异常后,对检验过程进行核查,确保了结果的准确性后主动与临床沟通,结合患者情况和实验室数据综合分析,避免患者进行过多的检查,为疾病的诊断提供有力的诊疗依据。 【参考文献】 [1] 田秦杰,黄尚志,葛秦生.雄激素受体异常与雄激素不敏感综合征[J].中华医学遗传学杂志(2期):95-97. [2] 沈永年,罗小平.儿科内分泌遗传代谢性疾病诊疗手册[M].上海科学技术文献出版社,2010. [3] Ahmed SF, Achermann JC, Arlt W, et al. UK guidance on the initial evaluation of an infant or an adolescent with a suspected disorder of sex development. Clin Endocrinol (Oxf) 2011; 75:12. [4] Doehnert U, Bertelloni S, Werner R, Dati E, Hiort O. Characteristic features of reproductive hormone profiles in late adolescent and adult females with complete androgen insensitivity syndrome. Sex Dev. 2015;9(2):69-74. [5] Rey RA, Belville C, Nihoul-Fékété C, et al. Evaluation of gonadal function in 107 intersex patients by means of serum antimüllerian hormone measurement. J Clin Endocrinol Metab. 1999;84(2):627-631. [6] 中国医师协会生殖医学专业委员会. 抗米勒管激素临床应用专家共识(2023年版)[J]. 中国实用妇科与产科杂志,2023,39(4):431-439. [7] 孙朋星,谭爱丽,洛若愚.MRKH综合征治疗进展[J].中国计划生育和妇产科,2022,14(05):47-49. [8] 郁琦,王含必,何方方.17α羟化酶缺乏症13例临床分析[J].中国实用妇科与产科杂志,2004,(11):19-21. [9] 马若骛,郭雅彬,张清学等.46,XY女性性发育异常临床分析[J].中华妇幼临床医学杂志(电子版),2022,18(01):61-66. [10] 聂应玲,朱岷.类固醇5α-还原酶2缺乏症研究进展[J].儿科药学杂志,2023,29(05):53-57. [11] Deans R, Creighton SM, Liao LM, Conway GS. Timing of gonadectomy in adult women with complete androgen insensitivity syndrome (CAIS): patient preferences and clinical evidence.Clin Endocrinol (Oxf). 2012;76(6):894-898. |
Copyright © 2015-2026 杭州宇翼科技有限公司 丨 Discuz! X3.5 丨增值电信业务经营许可证:浙B2-20190572丨浙ICP备18026348号-1丨浙公网安备33010802009352号