|
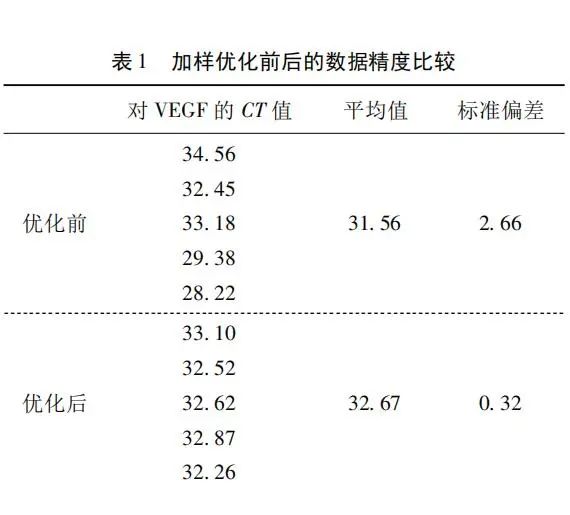
实时荧光定量 PCR(qPCR)自问世以来,就受到了广大生命科学实验工作者和医院检验工作者的极大关注,使得该技术得到了很快的普及。虽然qPCR技术目前应用方向十分广泛,但是仍碰到很多用户在实验中存在着由于不当操作而数据精度不高、重复性差、可信度差等问题。 引物的操作技巧 使用引物设计软件进行设计时,在操作界面上设定相应的参数,扩增子参数一般设定范围为80 ~ 200bp,扩增子长度越短,扩增效率越高,可选择设定在110bp,110bp在后期的扩增中仅用10s,其余的参数均默认,软件即给出引物列表,一般选用评分高或者靠前的,因为评分越高或越靠前的引物对形成引物二聚体的可能越小。 引物一般由试剂公司合成,为干粉状态,在溶解前,最好用高速离心机低温快速离心30s,使得引物干粉汇聚到管底,避免损失引物。引物收到后,需要先摸索引物的退火温度和终浓度,以达到较理想的qPCR扩增效果,其次测定引物的扩增效率,通常在非正式实验时,假定引物的扩增效率为100%,扩增产物在指数期呈现2n的增长。 在正式实验时,考虑到引物的扩增效率实际不是100%,所以需要测定引物的扩增效率以修正结果。一个测定引物扩增效率的简单方法如下: 以cDNA样品或标准品为模板,依次稀释 10*1、10*2、10*3、10*4、10*5、10*6 倍,采用前期摸索好的qPCR扩增条件,得到6个Cq值,然后以log(模板浓度)为X轴变量,以Cq值为Y轴变量作回归直线图,得到直线的斜率,扩增效率 = 10( -1 /斜率) - 1,整个数据计算可以在Excel中完成,也可在qPCR软件中直接得出。 反应体系配制技巧 正式实验时,每个样本至少3个重复,为了消除各重复之间的系统误差,将所需的所有试剂和模板加在同一个PCR管中,充分涡旋混匀后,再分装到3个重复孔中,这样可有效减小系统误差。 另外,一些实验者为了节省试剂,采用10μL反应体系,我们认为这是不可取的,因为体积太小,加样误差会大,这样做得不偿失,建议使用20μL以上的反应体积。如上操作,可以在较大程度上消除由于加样不准造成的系统误差。 如表1所列,“优化前”是5管中分别依次加入模板和各反应试剂,最后得到的Cq值标准偏差为2.66;“优化后”是使用模板和各反应试剂的混合液,然后分装至5管中,最后得到的Cq值标准偏差为0. 32;由上述可见,数据精度得到极大的改善。
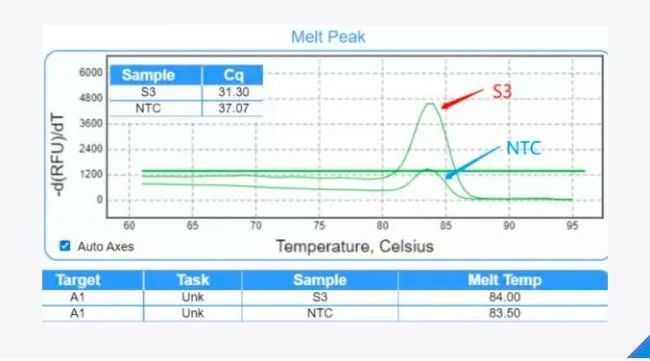
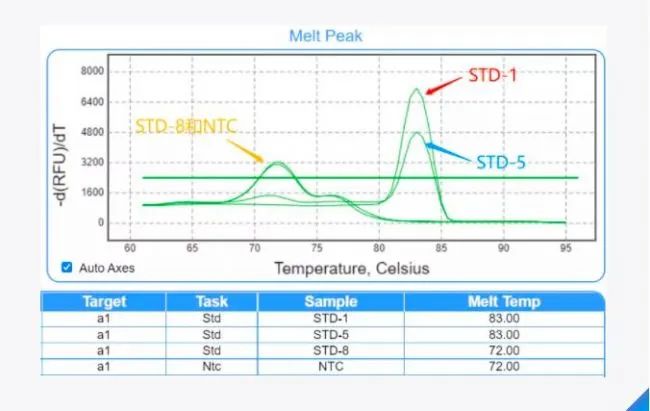
阴性对照的技巧 阴性对照一般是指用水代替模板的反应孔,体系中除了模板,包括了其他的反应组分。由于qPCR的高灵敏性,阴性对照有出现扩增信号的情况,那么在针对这类问题时,我们需要判断其原因所在。 使用染料法进行qPCR实验时,首先查看阴性对照的溶解曲线主峰是否跟阳性孔的主峰位置一致。如果一致,要看两者Cq相差多少,比如,针对某一种引物的实验样品Cq值为28. 5,阴性对照的Cq值为33. 6,由于Cq值相差大于5,如果按95%的扩增效率计算,两者模板量相差28倍之多,对分析数据影响不大,所以实验样品的Cq值还是可以采用的; 但是,如果两者的Cq值仅相差 1~2,这说明阴性对照中有较大的模板污染,则需要考虑更换试剂或引物,分区加样,重新实验。如果不一致,阴性对照主峰对应的Tm值小于阳性孔的Tm值,则可能是引物性能不好,无模板或使用低浓度模板下会出现引物二聚体,这种情况则需要考虑更换引物,保证引物特异性以及扩增效率。
▲ 图1:溶解曲线主峰位置基本一致时,S3是实验样品,NTC是阴性对照, 两者Cq相差大于5,实验样品的Cq值还是可以采用的。
▲ 图2:溶解曲线主峰位置不一致时,STD-1是高浓度样品,STD-5是中浓度样品, STD-8是低浓度样品,NTC是阴性对照。随着模板浓度降低逐渐出现引物二聚体, 使用TaqMan探针法进行qPCR实验时,由于探针法的高特异性,如果阴性对照出现扩增信号,很大可能是模板污染或气溶胶污染导致,操作时注意分区加样,避免污染。 结果分析的技巧 进行相对定量分析时,最常用到的是 2-ΔΔCq法,但是这种方法比较粗放,因为它的前提是假定所有引物的扩增效率都为100%。更为准确的定量则需要考虑不同引物扩增效率的差别。可首先用梯度稀释的模板测试各引物的扩增效率E,获得实际扩增效率之后,后续实验只需要在软件中直接输入扩增效率E值,即可计算更精准的相对定量值。 本文编辑:小珍 |
Copyright © 2015-2026 杭州宇翼科技有限公司 丨 Discuz! X3.5 丨增值电信业务经营许可证:浙B2-20190572丨浙ICP备18026348号-1丨浙公网安备33010802009352号