|
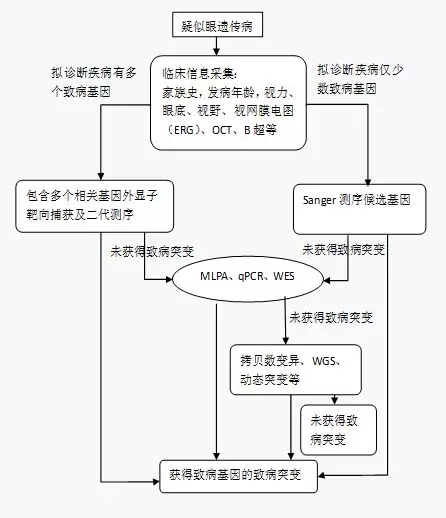
中国眼遗传病诊疗小组 中国眼科遗传联盟 通信作者:睢瑞芳, Email:hrfsui@163.com DOI:10.3760/cma.j.issn. 2095-0160.2018.07.001 基金项目:中国医学科学院医学与健康科技创新工程经费资助项目 (2016-I2M-1-002); 国家重点研发计划(2016YFC0901500) 眼遗传病是一组由于基因缺陷导致的眼部疾病。2018年4月27日在人类孟德尔遗传在线数据库(Online Mendelian Inheritance in Man,OMIM)(http://www.omim.org/)中以“eye”作为关键词搜索疾病表型可得到440种疾病条目。临床常见的眼遗传病有视网膜变性、先天性青光眼、先天性白内障、遗传性视神经病变、先天性眼外肌异常及累及眼部的一些综合征等。遗传方式包括常染色体显性遗传、常染色体隐性遗传、X性连锁遗传、双基因遗传及线粒体遗传等。此外,眼遗传病同样存在着等位基因异质性和基因座异质性。同时,不同地域、不同民族之间的基因变异和表型均存在着较大的差异,这些因素为眼遗传病的临床诊断和分子检测带来了巨大的挑战。随着人类基因组参考序列的完成、基因芯片和高通量测序等技术的问世以及生物信息技术对海量生物数据高效分析和处理技术的发展,近几年来单基因遗传病的分子诊断效率迅速提升,技术方法取得了很大的突破,为患者和临床医师的遗传咨询提供了技术保障,更为将来的基因治疗奠定了基础。目前,在众多基因组技术中二代测序(next-generation sequencing,NGS)技术尤以其特有的优势在包括眼遗传病在内的单基因遗传病的研究中发挥着重要作用,并越来越多地用于眼遗传病的分子检测。然而,由于基因检测技术的应用和方法选择在技术层面有一定的难度,更由于眼遗传性疾病有较大的基因突变异质性和临床表型异质性,在临床实践中我们发现对NGS的应用存在偏差,给遗传性眼病诊断结果和患者成本-效益带来一定的问题。为规范基因检测在眼遗传病分子诊断中的应用,我们组织了有关专家根据目前我国的实际情况,在深入分析及了解各种遗传性眼病表型的复杂性和致病基因变异复杂性的基础上进行反复讨论,提出眼遗传性疾病基因分子诊断规范化推荐意见,以供眼科相关临床人员和实验室检测人员在实践中参照应用。 本共识专家组成员由国内外人类眼遗传病专家和全国眼遗传病研究专家组成。共识的制定基于人类遗传性疾病基因分子诊断技术方法的科学性和适用范围,总结相关的推荐意见和观点,注重眼遗传性疾病基因分子诊断的临床实践可操作性和指导性,重点回答以下重要问题:(1) NGS的技术特点、适用范围及其优势和潜在局限性。(2)眼遗传性疾病如何遵循基因检测流程。(3)如何根据眼遗传疾病的临床特征合理选择基因检测方法。(4) 眼遗传病基因分子检测的目的及意义。 1 NGS技术及其应用 使用NGS技术可同时进行大量基因序列的平行测序。NGS分为靶向基因测序(target gene sequencing,TGS)(或称panel测序)、全基因外显子组测序(whole exome sequencing,WES)和全基因组测序(whole genome sequencing,WGS)。Panel测序和WES的基本流程为目标区域基因片段的获得和富集、对捕获片段的扩增和高通量测序、生物信息学分析及验证,最后确定致病突变。WGS是对全基因组的序列进行测序,故没有捕获这一步骤,可直接将基因组片段打断后进行高通量测序。这3种方法原理相似,但又有各自的特点。此外成本还存在差异,因而在应用范围上也各有优势。目前panel测序和WES广泛用于单基因遗传病的分子检测。 NGS技术对检测人员、实验室标准、试剂及项目选择、实验室质量管理等均有严格的要求,具体可参照《中华病理学杂志》在2017年3月发表的“临床分子病理实验室二代基因测序检测专家共识”。临床基因检测报告作为连接受检者、实验室技术人员和临床医师的重要依据,其内容在国内有也有了行业共识,具体可参考2018年2月《中华医学遗传学杂志》刊出的“临床基因检测报告规范与基因检测行业共识探讨”。该文对遗传病基因检测报告的原则、规范化和标准化提出了建议。结合基本规范,针对眼遗传性疾病已知的相关致病基因采用相应的优化检测模块,选择对外显子及已知内涵子突变全面覆盖的检测方法是提高检测结果可靠性的关键。 每种技术的临床应用过程中都存在不足,NGS也存在着一些缺点:(1)测序错误率高于传统的一代测序技术,需要增加测序深度、提高覆盖率等方法来补偿。(2)由于技术局限,对基因组鸟嘌呤和胞嘧啶所占比率高(GC含量)的目标区域捕获率达不到100%。(3)如果测序片段较短,高重复区域检测准确度下降。(4)对拷贝数变异(copy number variants,CNVs)、动态突变、复杂结构重排等变异类型的检测存在局限性。总之,在了解NGS优缺点的基础上,使其在临床单基因遗传病的分子检测中得到更合理的应用是目前基因检测过程中检测人员和临床医师面临的挑战[1]。 2眼遗传病基因检测流程 依据眼遗传病的临床特征选取单个或多个候选基因进行检测,应保证检测结果可靠,并遵循在选择检测方法时注重时效和减少费用的原则。目前致病基因的检测方法多种多样,包括Sanger测序法、实时荧光定量PCR(quantitative PCR,qPCR)、多重连接探针依赖的扩增技术(multiplex ligation dependent probe amplification,MLPA)、DNA微阵列、NGS等。其中常用的NGS技术获得的基因位点改变结果的可靠性需要通过Sanger测序加以验证(图1)。
图1 眼遗传病基因检测流程ERG:视网膜电图;MLPA:多重连接探针依赖的扩增技术;qPCR:实时荧光定量PCR;WES:全基因外显子组测序;WGS:全基因组测序 3 常见眼遗传病及其临床和基因特征 3.1 视网膜色素变性 3.1.1视网膜色素变性的临床特点 视网膜色素变性(retinitis pigmentosa,RP)是常见的一类遗传性致盲视网膜变性疾病,一般情况下视杆细胞最早出现功能受损且最为严重,同时或随后并发视锥细胞功能异常以及视网膜色素上皮细胞的损害。临床表现为夜盲、周边视野缩窄和视力下降,最终成为法定盲[2]。典型眼底改变包括视盘蜡黄、视网膜血管变细、中周部视网膜椒盐样改变和/或骨细胞样色素沉着。不同遗传方式、不同病因、不同年龄的RP患者其临床表现差异巨大。部分患者在疾病早期缺乏经典的眼底表现而易与其他眼病混淆;RP病变晚期及部分类型RP的早期病变还会合并黄斑病变;有些基因型改变的患者还存在特殊的眼底改变;对于伴RP的相关综合征患者,当其他器官病变特征或相关特征缺乏、不典型或未留意时,在临床上也可能诊断为单纯性RP。典型的RP患者借助于患者的主诉并结合眼底改变即可确诊。重要的辅助检查包括视网膜电图(electroretinogram,ERG)、视野和基因检测。荧光素眼底血管造影(fluorescein fundus angiography,FFA)、眼底自发荧光、光相干断层扫描(optical coherence tomography,OCT)及其他针对性检查有助于各种类型RP的鉴别。 3.1.2 RP相关的致病基因 RP具有高度的遗传异质性,已至少有87个基因可以导致RP(参考RetNet网站:https://sph.uth.edu/retnet/及根据网站数据摘录的表1)[2-6]。此外,伴RP的综合征,如Bardet-Biedl综合征、Usher综合征及其他RP相关疾病基因的突变也有可能导致RP或类似RP的表现。这些基因的突变可分别导致常染色体显性遗传、常染色体隐性遗传或X性连锁遗传RP,部分基因的突变还可以导致其他类型的视网膜变性[7-9]。大约60%的RP患者可以在这些基因中检测到致病突变基因[3,10-11],60%以上的突变集中于6个基因,即CYP4V2、RHO、USH2A、RPGR、CRB1和RP2[3]。报道较多的致病突变基因可作为突变的鉴定依据。对一些突变报道很少的基因进一步开展遗传学研究很有必要,对其临床基因检测要特别谨慎。已报道的部分RP基因也可能是错误的,尤其是一些有争议的或突变报道极少的基因。并不是所有明确的RP致病基因突变都是致病的,包括少数既往反复报道的、或者在人类基因突变数据库(Human Gene Mutation Database,HGMD)(http://www.hgmd.cf.ac.uk/ac/index.php)中记录的突变基因。在RP患者突变检测的同时分析特定突变在家系成员(尤其是父母)中的存在情况及其表型、对比特定类型突变在本民族人群及各数据库的频率、单个基因变异的系统梳理等均有助于提高临床基因检测的可靠性。除了极少数几个基因外,目前大多难以通过特征性表型如特异眼底改变来明确致病基因,因而对RP患者进行基因检测通常需对一组或全部相关基因进行系统分析。需要特别指出的是,由于RPGR ORF15区域的高度重复性,不易为大多数二代捕获测序覆盖,造成假阴性。鉴于RPGR基因ORF15突变高发的情况,有必要针对该区域采取优化捕获。 3.2视锥细胞或锥杆细胞营养不良 3.2.1视锥细胞或锥杆细胞营养不良的临床特点 目前我国尚缺乏视锥细胞或锥杆细胞营养不良(cone and cone rod dystrophy,CRD)的流行病学调查资料,欧洲的CRD发病率为1/40 000[12-13]。CRD表现为儿童期或成年早期进行性视力下降、畏光和色觉异常。疾病早期眼底检查接近正常,或仅有黄斑区色素不均或不同程度的黄斑区萎缩。病变晚期出现整个视网膜脉络膜萎缩及骨细胞样色素沉着。疾病早期视野即可呈现中心暗点。病变早期ERG为视锥细胞功能受损。随疾病的进展可逐步发展为视锥、视杆细胞功能的损伤。此外,眼底自发荧光、OCT、FFA可对CRD的诊断提供有用信息。 3.2.2 CRD的遗传特点 CRD的遗传方式有常染色体显性遗传、常染色体隐性遗传和X性连锁遗传。目前已确定的单纯性CRD致病基因有30余种,其中ABCA4为常染色体隐性CRD的主要致病基因[14],GUCY2D是常染色体显性CRD的主要致病基因[15]。 3.3黄斑营养不良 3.3.1黄斑营养不良的临床特点 黄斑营养不良(macular dystrophy,MD)主要包括青少年黄斑营养不良/眼底黄色斑点症(Stargardt disease/fundus flavimaculatus, STGD1)、卵黄样黄斑营养不良(Best vitelliform macular dystrophy,BVMD)及常染色体隐性卵黄样变(autosomal recessive bestrophinopathy,ARB)等。MD多由于青少年视力进行性下降、色觉异常和中心暗点等引起注意。在疾病的不同阶段眼底可出现相应的特征性改变,如黄斑区特征性黄色斑点或卵黄样外观。ARB患者可伴有闭角型青光眼。STGD早期ERG各波形正常,随着疾病的进展逐渐出现视锥细胞或视锥细胞、视杆细胞功能异常,表现为明视ERG a、b波的异常或最大混合光反应、暗视ERG和明视ERG a、b波的明显异常。BVMD和ARB患者眼电图(electro-oculogram, EOG)Arden 比值通常<1.5。眼底自发荧光、OCT及FFA对疾病的诊断有一定帮助。 3.3.2 MD的遗传特点 MD的遗传方式有常染色体显性遗传、常染色体隐性遗传和X性连锁遗传。目前已确定的单纯性MD致病基因有20余种,其中ABCA4是STGD1的唯一致病基因[16],BEST1是BVMD的致病基因[17]。 3.4 Leber遗传性视神经病变 3.4.1Leber遗传性视神经病变的临床特点 欧洲Leber遗传性视神经病变(Leber’s hereditary optic neuropathy,LHON)的发病率为1/31 000~1/50 000[18-19],我国目前尚无LHON相关的流行病学调查资料。无痛、无诱因地双眼先后或同时突发性视力下降是LHON的主要特征。约98%患者视力可降至0.1,但少有全盲者,部分患者视力可自行恢复。LHON可分为急性期和慢性期。LHON发病年龄从几岁到几十岁,男性青壮年者多发,约50%的男性突变携带者和10%的女性携带者会发病。多数患者为急性发病,之后双眼相继视力下降者约75%,双眼同时发生视力障碍者约25%,从视力下降到视力严重受损一般不超过8周。LHON急性期可见视网膜血管变形及充血、视神经纤维层水肿、视网膜动静脉迂曲扩张等, 但约20%的患者无上述表现。LHON的视野异常呈多样性,中心暗点和旁中心暗点者多见。LHON的色觉障碍常为后天获得性,红绿色盲多见,病情好转后色觉障碍也随之改善。家系中未发病者如有色觉障碍应定期随访。视觉诱发电位(visual evoked potential,VEP)检查有助于了解亚临床型或隐匿型LHON患者的视功能状况。 3.4.2 LHON遗传特点 LHON通过线粒体属母系遗传,主要为男性发病,但未见其直接遗传后代者。女性为遗传基因携带和传递者,但本身很少发病[20]。母亲将线粒体DNA传递给子女,但只有女儿将线粒体DNA传递给下一代。目前已发现18种基因突变与LHON有关,常见的3种为G11778A、G3460A和T14484C,90%以上的患者为此3种基因突变[21]。LHON属典型线粒体遗传性眼病,一代测序进行mtDNA突变筛查对大部分患者即可确诊,是基因诊断的第一步。未检测到突变基因时应排除假阴性结果(低于15% Sanger 测序可能测不出来)。排除假阴性结果后,需进行线粒体DNA环检测。 3.5常染色体显性遗传性视神经萎缩 3.5.1常染色体显性遗传性视神经萎缩的临床特点 常染色体显性遗传性视神经萎缩(autosomal dominant optic atrophy,ADOA)的发病率为1/10 000~50 000[22],主要表现为不同程度的视力下降,多在儿童期隐匿发病。ADOA的色觉障碍主要为蓝黄色觉异常,视野缺损,通常表现为中心盲点性暗点。眼底检查可见视盘颞侧苍白,病理改变主要表现为视网膜神经节细胞(retinal ganglion cells,RGCs)凋亡和视神经纤维丢失。图形VEP记录不到或峰时延长,图形ERG N95波和P50波振幅比值降低。OPA1基因突变携带者OCT检查提示神经纤维层和RGCs层变薄。 3.5.2 ADOA的遗传特点 ADOA是常染色体显性遗传性视神经疾病中最常见的一种,外显率为40%~90%[23],可表现为一种独立的疾病,也可伴不同程度的听力下降、白内障、眼外肌麻痹、上睑下垂等。基因检查显示突变侯选位点包括OPA1(3q28-29)、OPA3(19q13.2-13.3)、OPA4(18q12.2-12.3)和OPA5(22q12.1-13.1)等,其中OPA1与
Copyright © 2015-2026 杭州宇翼科技有限公司 丨 Discuz! X3.5
丨增值电信业务经营许可证:浙B2-20190572丨浙ICP备18026348号-1丨浙公网安备33010802009352号
|