伴随诊断(Companion Diagnostic ,CDx),对于其伴随药物的安全性及有效性非常重要,用于检测患者特定生物标志物,并识别出哪些患者能从药物治疗中明显获益或副作用风险增加。2014年,美国Food and Drug Administration(FDA)发布的《In Vitro Companion Diagnostic Devices》首次明确定义了伴随诊断。此后日本Pharmaceuticals and Medical Devices Agency(PMDA)、欧盟European Medicines Agency(EMA)相继发布了相关指南并定义了伴随诊断,中国National Medical Products Administration(NMPA)在2020年发布的《已上市抗肿瘤药物的伴随诊断试剂临床试验指导原则(征求意见稿)》中也初步定义了伴随诊断。表1比较了FDA、PMDA、EMA、NMPA对于伴随诊断的定义[1,2,3,10]:

表1. FDA、PMDA、EMA有关伴随诊断定义的比较[1,2,3,10]
1998年,FDA批准了HercepTest用于识别能从曲妥珠单抗(Trastuzumab)治疗中获益的人群,这是第一个FDA批准的伴随诊断,截止2021年6月FDA已经批准了45个伴随诊断,主要基于免疫组织化学(Immunohistochemistry,IHC)、荧光原位杂交(Fluorescence In Situ Hybridization,FISH)、显色原位杂交(Chromogenic In Situ Hybridization,CISH)、聚合酶链式反应(Polymerase Chain Reaction,PCR)、下一代测序(Next Generation Sequencing,NGS)等方法开发,表2列举了基于这些方法开发的伴随诊断[4,5]:
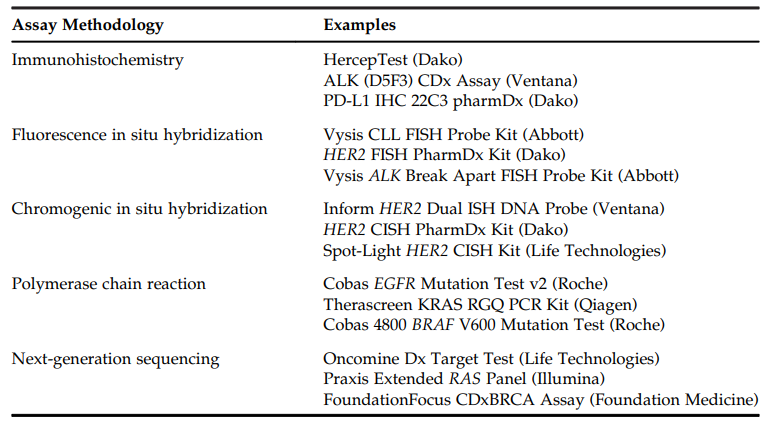
表2. FDA批准的伴随诊断方法学示例[4]
图1展示了FDA历年批准的伴随诊断数量(按方法学分类统计):
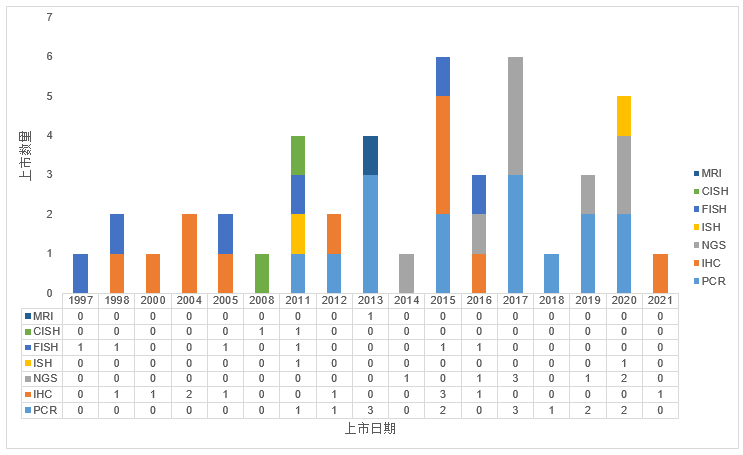
图1. FDA历年批准的伴随诊断数量(按方法学分类统计)【5】
伴随诊断的开发方法主要有同步(Co-development)、桥接(Bridging)、跟随(Follow-on)三种。FDA鼓励伴随诊断企业与药物研发企业密切合作,通过同步的方式可更经济地实现伴随诊断及其伴随药物的同时获批(Co- approval)。如果申请注册的伴随诊断(Candidate IVD Companion Diagnostic)与临床试验中用于主要疗效试验(Major Efficacy Trial)的临床试验检测(Clinical Trial Assay,CTA)不一样,FDA要求通过桥接研究的方式,分析原始临床试验样本来证明两种方法具有非常相似的性能特征。如果伴随诊断企业无药企合作伙伴或者最初临床试验中使用的样本无法获得,可使用跟随的方式,Meijuan Li等在《Statistical methods for clinical validation of follow-on companion diagnostic devices via an external concordance study》中对跟随研究面临的问题及解决方案作了详细的介绍[6]。图2比较了同步、桥接、跟随三种伴随诊断开发方法的特点:
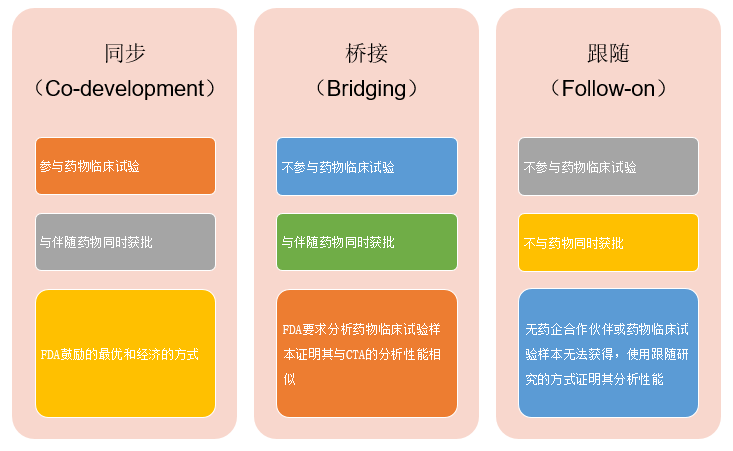
图2. 同步、桥接、跟随三种伴随诊断开发方法的特点
如图3所示,由伴随诊断企业负责的生物标志物研究及伴随诊断开发,可在药物研发的不同阶段进行:
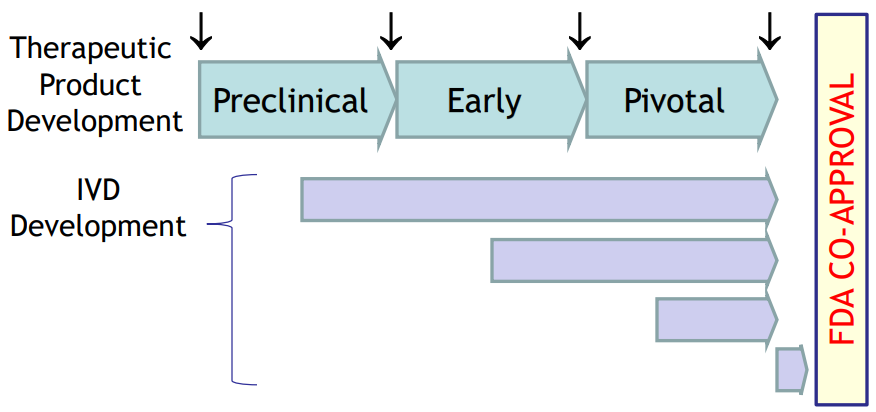
图3. Biomarker discovery (↓) and test development can occur at any point during the therapeutic product development process【11】.
2014年,Nobuyuki Hanamura等在Personalized Medicine上发表了《Global development strategy for companion diagnostics based on the usage and approval history for biomarkers in Japan, the USA and the EU》,总结了伴随诊断的开发策略,以及伴随诊断全球开发过程中面临的主要挑战[7]。如图4所示,伴随诊断开发策略主要有四种方式:
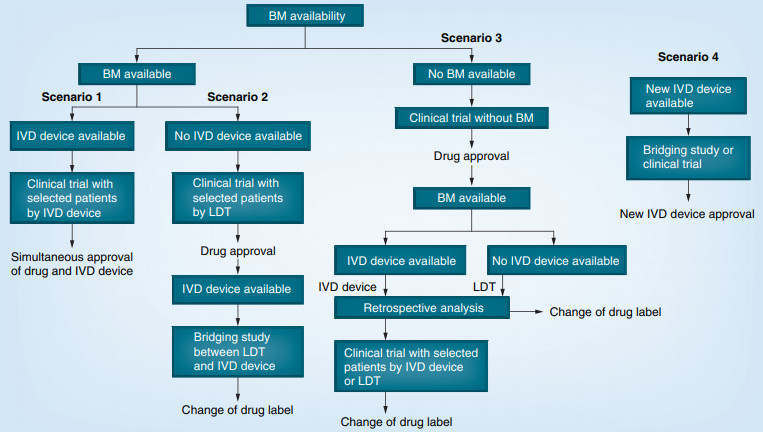
图4. 伴随诊断开发策略[6]
1.策略一:如果药物开发(Drug Development)初期,用于患者筛选(Patient Selection)的生物标志物(Biomarker,BM)是明确的,临床试验应该对生物标志物阳性(Biomarker Positive Cohorts)、阴性(Biomarker Negative Cohorts)的两个队列进行研究,并且有已经获批的体外诊断(In Vitro Diagnostic,IVD)用于该生物标志物的检测时,那么应该采用共同开发(Co-development)的策略。共同开发的伴随诊断举例:①Crizotinib and the Vysis ALK Break Apart FISH kit (Japan, USA)②Cetuximab and EGFR PharmDx (Japan, USA)③Panitumumab and EGFR PharmDx (Japan, USA)④Panitumumab and the TheraScreen KRAS mutation kit (Japan)2.策略二:当没有批准的IVD用于该生物标志物的检测时,应该使用实验室自建检测(Laboratory-Developed Tests,LDTs)进行患者筛选,如果药物开发后期有IVD获批,应该进行桥接研究(Bridging Study)验证LDT与IVD具有非常类似的检测性能(Very Similar Performance Characteristics)。例如曲妥珠单抗在乳腺癌临床试验时使用临床试验检测(Clinical Trial Assay,CTA)进行患者筛选,后来通过桥接研究证明了HercepTest与CTA检测结果的一致性。3.策略三:如果药物开发初期,用于患者筛选的生物标志物不明确,临床试验不需要进行基于生物标志物分组的队列研究。一旦生物标志物明确,应该进行回顾性分析(Retrospective Analysis)来评价生物标志物。如果生物标志物阳性队列的有效性或安全性(Efficacy/Safety)优于阴性队列,那么药物说明书(Drug Label)中应该增加生物标志物的相关信息。例如KRAS mutation kits和其伴随药物西妥昔单抗(Cetuximab),就是通过这种策略开发的。4.策略四:如果开发新的针对相同适应症的伴随诊断或对已有伴随诊断进行改进,应该与已有伴随诊断进行桥接研究,否则应该进行临床试验研究来验证其检测性能。HercepTest是第一个曲妥珠单抗的伴随诊断,后来Abbot和Dako通过这种策略开发了曲妥珠单抗的伴随诊断:①Pathvision HER-2 DNA Probe kit (Abbot, USA)②Dako Cytomation Her2 FISH PharmDx™ kit (Dako Cytomation,Denmark)伴随诊断的全球开发,可节约共同开发的周期及成本,同时能加快药物研发进展及提高成功率,但是需要考虑全球开发面临的独特挑战。Nobuyuki Hanamura深入分析了六个药物(Carbamazepine、Rasburicase、Abacavir、Panitumumab、Exemestane、Letrozole)的伴随诊断,这些药物在不同区域都获批了伴随诊断,但是伴随诊断检测的方法学及生物标志物有所差异,这些差异也是伴随诊断全球开发策略中需要重点关注的问题:
- 种族差异:由于种族的差异,不同地区人群中某个生物标志物的阳性比例不一样,这会导致不同地区选择不同的检测方法开发伴随诊断;
- 使用不同检测方法筛选患者:临床试验中,在不同地区或不同时间使用不同的方法筛选患者,这会导致不同地区检测的生物标志物不同。
如图5所示,虽然美国伴随诊断相关法规比较完善了,但是花了比较长的时间来探索与建立。2020年,CMDE先后发布了《已上市抗肿瘤药物的伴随诊断试剂临床试验指导原则(征求意见稿)》、《基于同类治疗药品的伴随诊断试剂说明书更新与技术审查指导原则(征求意见稿)》两份征求意见稿,同时也启动了征集伴随诊断试剂生产企业信息参与编制《伴随诊断试剂与抗肿瘤药物同步研发的临床试验技术指导原则》,足以说明CMDE非常重视伴随诊断相关法规的建立与完善,并进一步规范国内伴随诊断开发的合规性。
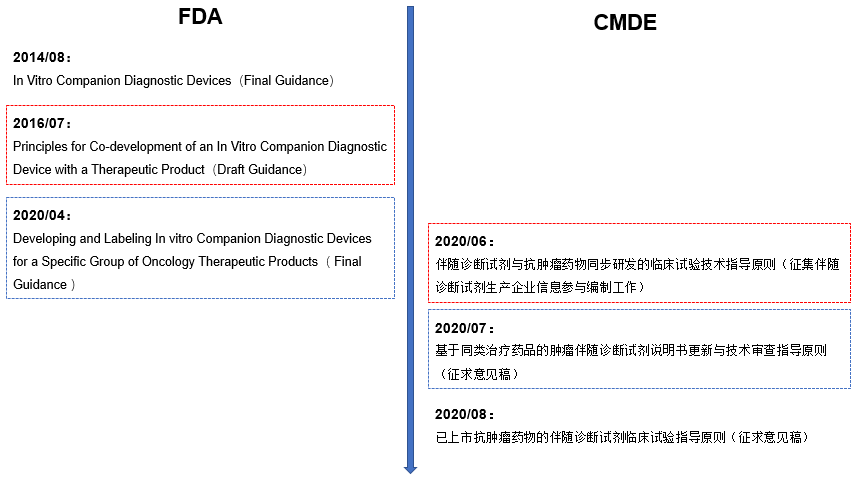
图5. 中美伴随诊断法规进展【8,9】
迈杰转化医学作为国内精准诊断整体解决方案的领导者,致力于解决精准医疗药物研发及患者用药痛点,围绕生物标志物研究、伴随诊断开发,建立了完善的核酸组学、蛋白组学、细胞组学技术平台。为创新药企开展全球多中心临床试验研究,提供中心实验室检测及伴随诊断开发服务,同时解决伴随诊断全球开发面临的问题。
更多伴随诊断信息,欢迎咨询MARKETING@MEDxTMC.com。
参考资料:
Food and Drug Administration,In Vitro Companion Diagnostic Devices:Guidance for Industry and Food and Drug Administration Staff,Document issued on: August 6, 2014.
Pharmaceuticals and Medical Devices Agency,Notification on Approval Application for In Vitro Companion Diagnostics and Corresponding Therapeutic Products,PFSB/ELD Notification No. 0701-10 July 1, 2013.
European Medicines Agency, Regulation (EU) 2017 746 on In-Vitro Diagnostic Devices, Official Journal of the European Union 5.5.2017.
Jan TrøstJørgensen, et al. Companion and Complementary Diagnostics: From Biomarker Discovery to Clinical Implementation, ISBN: 978-0-12-813539-6.
Food and Drug Administration, List of Cleared or Approved Companion Diagnostic Devices (In Vitro and Imaging Tools).
Li,Meijuan,et al. Statistical methods for clinical validation of follow-on companion diagnostic devices via an external concordance study, Statistics in Biopharmaceutical Research: ISSN: (Print) 1946-6315.
Nobuyuki Hanamura,et al. Global development strategy for companion diagnostics based on the usage and approval history for biomarkers in Japan, the USA and the EU, Personalized Medicine (2014) 11(1), 27–40.
https://www.fda.gov/.
https://www.cmde.org.cn/.
国家药品监督管理局,《已上市抗肿瘤药物的伴随诊断试剂临床试验指导原则(征求意见稿)》.
CDRH Learn/Principles for Codevelopment of an In Vitro Companion Diagnostic Device with a Therapeutic Product: PowerPoint Presentation (fda.gov).



