|
质量是临床检验的根本,影响临床结果的判断和病人的治疗方案,并关乎病人的生命。高质量的产品可以确保临床诊断的准确性。 质量是国家之间竞争的重要因素,高质量产品的输出是国家走向世界先进水平的必由之路。 一、美国FDA 美国食品药品监督管理局(英语:U.S. Food and Drug Administration,缩写为FDA)为美国卫生与公众服务部直辖的联邦政府机构,其主要职能为负责对美国国内生产及进口的食品、膳食补充剂、药品、疫苗、生物医药制剂、血液制剂、医疗设备、放射性设备、兽药和化妆品进行监督管理,同时也负责执行公共健康法案(the Public Health Service Act)的第361号条款,包括公共卫生条件及州际旅行和运输的检查、对于诸多产品中可能存在的疾病的控制等等。 1) 政府专员办公室(OC) 2) 药品审评和研究中心(CDER) 3) 生物制品审评和研究中心(CBER) 4) 食品安全和应用营养中心(CFSAN) 5) 设备与放射健康中心(CDRH) 6) 兽药中心(CVM) 7) 国家毒理学研究中心(NCTR) 8) 监管事务办公室(ORA)
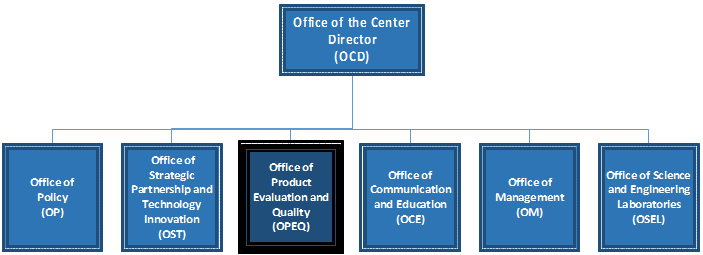
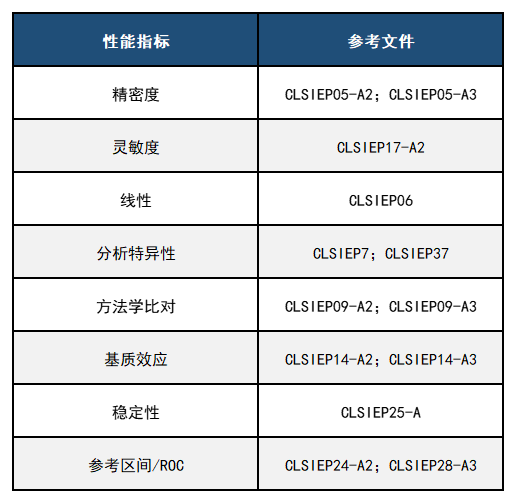
(1)成立了产品评估和质量办公室(OPEQ)–将合规办公室、设备评估办公室、监视和生物识别办公室以及体外诊断和放射健康办公室合并为一个专注于整个产品生命周期方法的超级办公室对医疗器械的监督。 (2)建立了政策办公室-包括两个团队,即指导立法和特别项目团队以及法规文件和特别项目团队。 (3)建立了战略合作伙伴关系和技术创新办公室–合并了科学和战略合作伙伴关系,数字健康、标准、健康信息学和创新团队。 (4)中心内重新调整的管理服务–确保CDRH中的管理职能得到最佳调整,结构化并提供出色的服务。 (5)简化了中心的沟通功能–将内部和外部沟通功能(包括CDRH ExecSec和议长联络员)组合到了沟通和教育办公室更名为沟通部,并建立了内部沟通处。 二、FDA 510(k) 1、产品分类《联邦食品、药品和化妆品法》的513节(FD&C ACT section 513),根据医疗器械的使用风险和可能产生的风险程度,将医疗器械划分为三个管理类别,采取不同程度的控制措施,根据美国医疗器械分类标准,医疗器械分类以“对产品的控制程度为基础”,为确保医疗器械产品的安全有效性,分为“基本控制”和“特殊控制”。产品被列为不同的类别,意味着其制造商向FDA申请获准上市的程序和提交资料将有所区别。(1) I类器械(一般控制)这类器械只需经过一般控制就可以确保其功效与安全性,如拐杖、眼镜片、胶布等。一般控制的内容包括:禁止粗制滥造及不当标示的产品销售、不良产品限制或禁止销售和使用的规定、有关通知消费者和修理、更换、补偿金等售后服务的规定、GMP要求和制造商,进口商及分销商的企业注册与产品登记等。划入II类、III类的器械同样要遵守以上的要求。2、上市前通知2) 相同用途、不同技术特征,但具有相同的安全性以及有效性,而且不会产生与对比器械(predicate device)不同的有关安全与有效性方面的问题。 1)申请函,此部分应包括申请人(或联系人)和企业的基本信息、510(K)递交的目的、申请上市器械的名称型号和分类资料、进行实质等效比较的产品(Predicate Device)名称及其510(K)号码; 2)目录,即510(K)文件中所含全部资料的清单(包括附件); 3)真实性保证声明,对此声明,FDA有一个标准的样本; 4)器材名称,即产品通用名、FDA分类名、产品贸易名; 5)注册号码,如企业在递交510(K)时已进行企业注册,则应给出注册信息,若未注册,也予注明; 6)分类,即产品的分类组、类别、管理号和产品代码; 7)性能标准,产品所满足的强制性标准或自愿性标准; 8)产品标识,包括企业包装标识、使用说明书、包装附件、产品标示等; 9)实质相等性比较(SE)摘要; 10)510(K)摘要或声明; 11)产品描述,包括产品的预期用途、工作原理、动力来源、零组件、照片、工艺图、装配图、结构示意图等; 12)产品的安全性与有效性,包括各种设计、测试资料; 13)生物相容性; 14)色素添加剂(如适用); 15)软件验证(如适用); 16)灭菌(如适用),包括灭菌方法的描述、灭菌验证产品包装和标识等。 4、申请流程1) 确认付款(FDA确认收到审核费,如确认收到审核费则继续受理工作):提交文件时要包括一份纸质档和电子档,以及内容一致性声明,FDA收到后会确认收没收到审核费。 2) 行政审核:FDA检查电子文件的格式是否符合要求,如不符合要求有最长180天的整改时间,FDA确认是否申请所有的要素都已经提供;如不符合要求有最长180天的整改时间。 3) 实质审核:FDA会进行最长60天的实质审核,实质审核结束后会做出决定发布或者交互审核。 4) 补充材料:按照FDA的要求准备补充材料,有最长180天的整改时间。 5) 交互审核:FDA会对补充材料进行审核,补充材料的审核时间根据情况有所差异。 三、产品性能评估要求 申请美国FDA认证过程中,需要提交技术文档,其中最为重要的是产品性能资料,其中需要包括精密度、灵敏度、线性、稳定性、分析特异性、方法学比对、参考区间等,尤其是参考区间需要考虑人种对项目的影响,需要在美国本地完成参考区间的建立。另外,方法学比对需要考虑至少三个实验室三台仪器的差异性,同时也需要在美国本地完成方法学比对测试。各类性能的评估需要严格按照美国CLSI EP指导文件的方案和标准,具体各性能参考的标准如下表1所示:
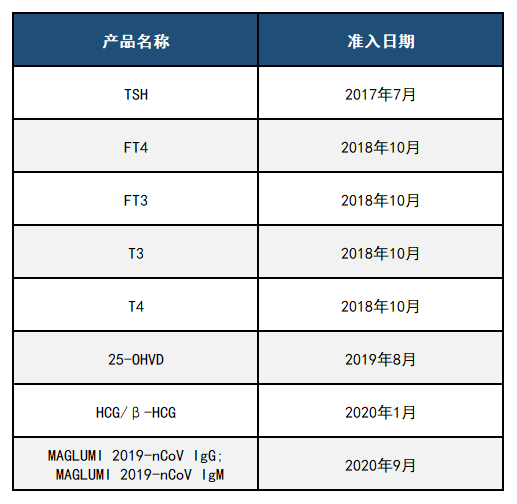
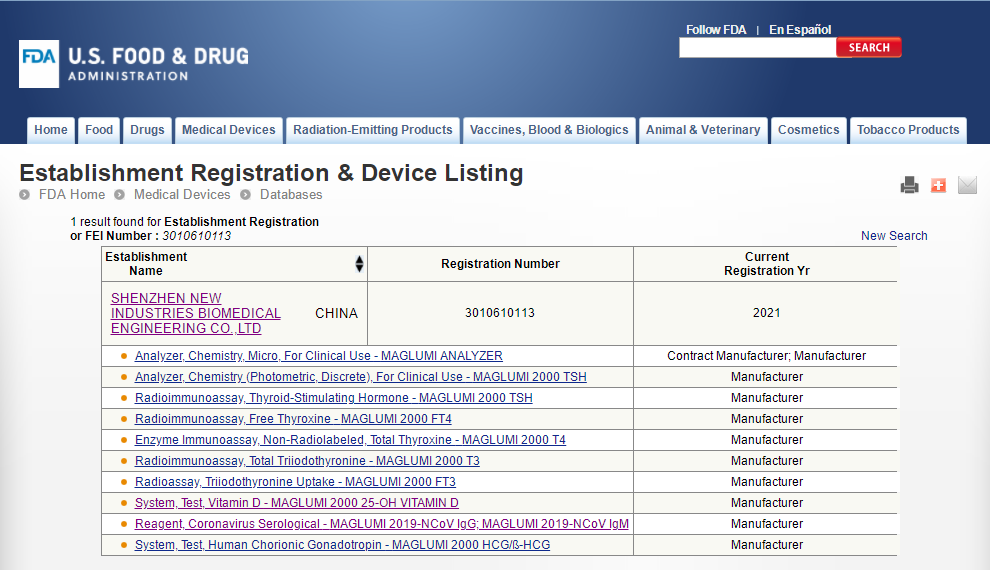
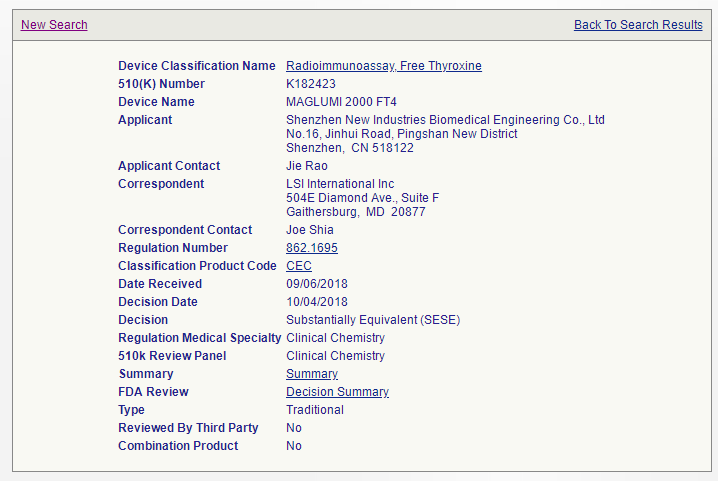
四、新产业FDA准入产品 新产业作为化学发光免疫分析领域的国际领先企业,一直秉承着“像捍卫生命一样捍卫产品质量”的崇高理念,将产品质量放在至高无上的地位。众多产品成功获得美国FDA市场准入,成为中国首家获得FDA准入的化学发光厂家,同时也是中国第一家丙肝试剂荣获欧盟最高标准CE ListA认证的化学发光厂家。 新产业参与了伯乐、朗道质控品赋值,产品远销全球150多个国家和地区。产品质量获得国际学术界、检验界的高度认同,用新产业产品在全球多种顶级杂志发表多篇高质量有影响的学术论文,例如Nature子刊Nature Communication(2021年影响因子14.919)、美国医学会杂志JAMA(2021年影响因子56.272)等。新产业全面参与并以高质量通过国家卫健委以及各省的室间质评。 新产业专注于体外诊断行业26年,目前已围绕主营产品建立了磁性微球、试剂关键原料、仪器研发及试剂研发四大技术平台,从原料、工艺、生产、质控、销售售后等建立了一系列系统完善的质量管理制度,每一个环节都严格遵守国际最高标准,确保了高质量的试剂产品。 新产业目前获得美国FDA准入的产品一共有9项。具体信息如下表2所列:
|
Copyright © 2015-2023 杭州宇翼科技有限公司 丨 Discuz! X3.5 丨增值电信业务经营许可证:浙B2-20190572丨浙ICP备18026348号-1丨浙公网安备33010802009352号