|
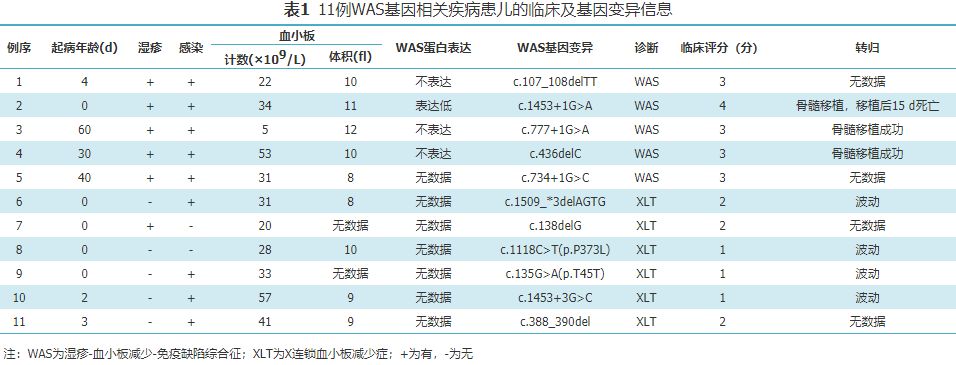
摘要 目的 探讨基因筛查用于早期识别新生儿WAS基因相关疾病的临床应用价值。 方法 2016年1月至2017年12月复旦大学附属儿科医院新生儿基因组计划纳入新生儿重症监护病房中的5 800例高危新生儿,采用高通量测序方法进行遗传病因的分析,检测到的11例携带WAS基因致病或可疑致病变异的新生儿。回顾性总结11例患儿(均为男性)临床特征,并结合基因变异特征,进行表型基因型分析。 结果 11例患儿中5例为湿疹-血小板减少-免疫缺陷综合征(WAS),6例为X连锁血小板减少症(XLT)。WAS患儿中2例在新生儿早期、3例在5~8周龄出现临床表现。3例XLT新生儿因其他疾病住院治疗,发现血小板减低。检测到WAS基因致病或可疑致病变异11个,其中7个为首次报道的致病或可疑致病变异,包括2个移码变异c.138delG和c.388_390del,4个剪接位点变异c.1453+1G>A、c.734+1G>C、c.135G>A和c.1453+3G>C,1个错义变异c.1118C>T。另外4个已报道致病变异是c.777+1G>A、c.107_108delTT、c.436delC和c.1509_*3delAGTG。3例患儿接受了造血干细胞移植治疗,2例治愈。 结论 WAS基因相关疾病在新生儿期缺乏特征性表现,新生儿期基因筛查有助于早期识别和干预该类疾病。 WAS基因相关疾病是由于WAS基因变异所致的一类原发性免疫缺陷病。根据临床表现可分为Wiskott-Aldrich综合征(Wiskott-Aldrich syndrome,WAS)、X-连锁血小板减少症(X-linked thrombocytopenia,XLT)、间歇性X-连锁血小板减少症(intermittent X-linked thrombocytopenia,IXLT)和X-连锁中性粒细胞减少症(X-linked neutropenia,XLN),均为X连锁隐性遗传。其中WAS是最常见的临床类型,WAS又称湿疹、血小板减少伴免疫缺陷综合征,以湿疹、血小板减少伴体积减小、反复感染、易合并自身免疫性疾病及恶性肿瘤为特征,因最先由Alfred Wiskott和Robert Aldrich发现而命名[1],发病率占出生男孩(1~10)/1 000 000[2]。该病发病年龄早,多发于新生儿期,绝大多数起病于6月龄内[3]。1994年,Derry等[4]通过定位克隆技术明确WAS基因变异为该病的根本原因。由于WAS基因变异所致疾病的临床表现差异大,很多患儿虽然起病年龄早,但诊断年龄滞后,平均确诊年龄为10.8月龄,严重影响患儿的预后。WAS患儿若不经造血干细胞移植治疗其生存期仅为15年[5],因此,早期诊断是改善预后的关键因素。为探讨早期诊断WAS基因相关疾病的方法,2016至2017年对参与新生儿基因组计划的患儿进行了回顾性分析,旨在探讨基因筛查用于早期识别新生儿WAS基因相关疾病的临床应用价值。 感染是导致新生儿死亡的重要原因。世界卫生组织(World Health Organization,WHO)数据显示,全球每年约有400万新生儿死亡,其中近1/3死于感染,感染已成为导致新生儿死亡的第二位因素(仅次于早产及其并发症)[7,8]。新生儿感染之所以死亡率高,除病原因素之外,免疫功能状态是重要影响因素。WAS基因位于X染色体短臂的Xp11.23-p11.22区域,包含12个外显子,编码由502个氨基酸组成的WAS蛋白[9]。WAS蛋白是一种富含脯氨酸的胞质蛋白,表达于各类血细胞和免疫活性细胞,维持各系血细胞生成、迁移以及调节免疫细胞正常活性[10]。WAS基因致病变异会导致WAS蛋白表达异常或缺失,进而影响血小板生成和聚集以及淋巴细胞迁移、信号传导和突触形成。 WAS基因相关疾病多起病于新生儿期,近年来有研究发现表型较重的WAS发病年龄较早(平均0.7月龄),而XLT发病年龄较晚(平均7.07月龄)[3]。湿疹、血小板减少和免疫缺陷是WAS的典型临床特征,但在新生儿期,常缺乏上述典型的三联征。本组11例有明确临床信息的患儿,起病年龄为0~8周龄,8例患儿起病于生后第1周,但其中3例就诊原因为其他疾病,入院后发现血小板减少;3例起病于5~8周龄。且首发的表现特征性不高,包括便血或湿疹或皮肤出血点,故而在疾病早期不足以引起新生儿科医师的重视。本组数据提示,WAS多症状较为严重,但也可在新生儿期后出现相应临床表现。 WAS基因相关疾病不能完全依靠于新生儿期的特征性表现进行临床诊断。目前已知WAS蛋白表达情况和疾病严重程度相关。WAS蛋白阳性患儿(一般表达减少)临床表型较轻为XLT;WAS蛋白阴性患儿(表达缺失或表达截短型WAS蛋白)临床表型较重为WAS[11]。本组4例WAS患儿检测了WAS蛋白表达情况,其中3例患儿蛋白不表达,1例患儿表达降低,支持既往研究结论[11,12]。这4例患儿均携带移码变异和剪接位点变异。对于新生儿期的轻度出血倾向表现,应警惕WAS基因变异的可能,早期通过蛋白水平、基因水平协助早期诊断。 基因筛查有助于早期识别WAS基因相关疾病,为治疗手段的进一步选择和预后判断提供证据。已知的WAS基因致病变异约有300种,包括错义变异、拼接位点变异、缺失变异、无义变异、插入变异及复合变异。本组5例WAS患儿具有较严重的表型,包括血小板减少、湿疹、感染,其余6例XLT患儿表型较轻。11种WAS基因变异包括7个新发变异和4个已知变异(其中1个为热点变异)。与以往报道相比,本组WAS患儿的基因变异情况相似,而XLT患儿的变异种类多样,并且剪接位点变异和移码变异占了大多数。目前报道的基因致病变异类型与疾病表型的关联主要为XLT或表型较轻的疾病多为错义变异,WAS或表型较重的疾病多为无义、缺失、插入、拼接位点变异[13,14]。值得注意的是,在2例患儿的11号外显子上检测到2个位置相近的剪接位点变异c.1453+1G>A和c.1453+3G>C,临床分别诊断为WAS和XLT,其中XLT患儿于5日龄检测到血小板减少,住院1个月余出院,对该患儿应密切随访表型便于尽早治疗。目前造血干细胞移植仍是临床根治WAS最有效的方法。本组3例WAS患儿接受了造血干细胞移植治疗,其中2例病情得到控制。 本研究采用的临床全外显子组测序的方法,在检测的5 800例患儿中,仅WAS基因变异这一种疾病就检出11例,远远超过整个人群的发生率,提示NICU住院患儿为高危人群,有必要进行基因检测,进行尽早干预,即可节省抗感染、生命支持等费用。本数据提示,依靠WAS基因变异的类型,有助于判断临床的分型,对于携带错义变异的患儿,其导致WAS的可能性相对较小。本研究补充丰富了WAS基因的致病变异谱,为临床早期遗传检测、临床干预和治疗WAS基因相关疾病提供了初步依据,也为家系提供遗传咨询,为致病变异携带者进行胚胎植入前诊断、产前检测提供了基础[15]。 本研究的不足,一方面由于此类罕见疾病,样本量比较小,需要不断扩充样本量从而形成更加具有说服力的结论;另一方面,需要不断完善新生儿期此类疾病的跟踪随访队列,对于此类遗传相关疾病的后期临床表现进行系统和完善的整理。 参考文献(略)
|
Copyright © 2015-2023 杭州宇翼科技有限公司 丨 Discuz! X3.5 丨增值电信业务经营许可证:浙B2-20190572丨浙ICP备18026348号-1丨浙公网安备33010802009352号